Biophysical and Computational Landscapes of Mycobacterial Pyrophosphatase: a Closer View of Drug Repurposing with Different Natural and Synthetic Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum Brent Elliot Wells
The University of Maine DigitalCommons@UMaine Electronic Theses and Dissertations Fogler Library 2003 Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum Brent Elliot Wells Follow this and additional works at: http://digitalcommons.library.umaine.edu/etd Part of the Biochemistry Commons Recommended Citation Wells, Brent Elliot, "Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum" (2003). Electronic Theses and Dissertations. 301. http://digitalcommons.library.umaine.edu/etd/301 This Open-Access Thesis is brought to you for free and open access by DigitalCommons@UMaine. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of DigitalCommons@UMaine. IDENTIFICATION AND PARTIAL CHARACTERIZATION OF A FAMILY OF PUTATIVE PALMITOYLTRANSFERASES IN DICTYOSTELIUM DISCOIDEUM BY Brent Elliott Wells B.S. University of Utah, 1994 A THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science (in Biochemistry) The Graduate School The University of Maine December, 2003 Advisory Committee: Robert E. Gundersen, Associate Professor of Biochemistry, Advisor Keith W. Hutchison, Professor of Biochemistry Mary Rumpho-Kennedy, Professor of Biochemistry LIBRARY RIGHTS STATEMENT In presenting this thesis in partial fulfillment of the requirements for an advanced degree at The University of Maine, I agree that the Library shall make it freely available for inspection. I further agree that permission for "fair use" copying of this thesis for scholarly purposes may be granted by the Librarian. It is understood that any copying or publication of this thesis for financial gain shall not be allowed without my written permission. -

Hras Intracellular Trafficking and Signal Transduction Jodi Ho-Jung Mckay Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2007 HRas intracellular trafficking and signal transduction Jodi Ho-Jung McKay Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Biological Phenomena, Cell Phenomena, and Immunity Commons, Cancer Biology Commons, Cell Biology Commons, Genetics and Genomics Commons, and the Medical Cell Biology Commons Recommended Citation McKay, Jodi Ho-Jung, "HRas intracellular trafficking and signal transduction" (2007). Retrospective Theses and Dissertations. 13946. https://lib.dr.iastate.edu/rtd/13946 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. HRas intracellular trafficking and signal transduction by Jodi Ho-Jung McKay A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Genetics Program of Study Committee: Janice E. Buss, Co-major Professor Linda Ambrosio, Co-major Professor Diane Bassham Drena Dobbs Ted Huiatt Iowa State University Ames, Iowa 2007 Copyright © Jodi Ho-Jung McKay, 2007. All rights reserved. UMI Number: 3274881 Copyright 2007 by McKay, Jodi Ho-Jung All rights reserved. UMI Microform 3274881 Copyright 2008 by ProQuest Information and Learning Company. All rights reserved. This microform edition is protected against unauthorized copying under Title 17, United States Code. ProQuest Information and Learning Company 300 North Zeeb Road P.O. -

Inorganic Pyrophosphatase Is a Component of the Drosophila Nucleosome Remodeling Factor Complex
Downloaded from genesdev.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Inorganic pyrophosphatase is a component of the Drosophila nucleosome remodeling factor complex David A. Gdula, Raphael Sandaltzopoulos, Toshio Tsukiyama,1 Vincent Ossipow, and Carl Wu2 Laboratory of Molecular Cell Biology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892-4255 USA The Drosophila nucleosome remodeling factor (NURF) is a protein complex consisting of four polypeptides that facilitates the perturbation of chromatin structure in vitro in an ATP-dependent manner. The 140-kD NURF subunit, imitation switch (ISWI), is related to the SWI2/SNF2 ATPase. Another subunit, NURF-55, is a 55-kD WD repeat protein homologous to the human retinoblastoma-associated protein RbAp48. Here, we report the cloning and characterization of the smallest (38 kD) component of NURF. NURF-38 is strikingly homologous to known inorganic pyrophosphatases. Both recombinant NURF-38 alone and the purified NURF complex are shown to have inorganic pyrophosphatase activity. Inhibition of the pyrophosphatase activity of NURF with sodium fluoride has no significant effect on chromatin remodeling, indicating that these two activities may be biochemically uncoupled. Our results suggest that NURF-38 may serve a structural or regulatory role in the complex. Alternatively, because accumulation of unhydrolyzed pyrophosphate during nucleotide incorporation inhibits polymerization, NURF may also have been adapted to deliver pyrophosphatase -
Type of the Paper (Article
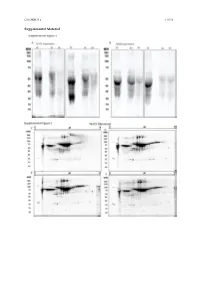
Cells 2020, 9, x 1 of 19 Supplemental Material Cells 2020, 9, x 2 of 19 Figure S1. Secretome enrichment: protocol optimization. 1D SDS-PAGE documentation of washing steps: Culture medium was substituted with FCS-free medium, which was changed every 2 h. The supernatants were then collected, and the proteins isolated and separated in 1D SDS-PAGE ((A) TK173 and (B) TK188). Proteins were stained with Flamingo fluorescent gel stain. Two-dimensional pattern of the proteins isolated from supernatant of TK173, (C) 2 h, (D) 4 h, (E) 6 h, and (F) 8 h after changing to FCS-free medium. (G) Cell secretome collected 24 h after elimination of the contaminating FCS-proteins with different washing steps. Proteins were stained with Flamingo fluorescent gel stain. Cells 2020, 9, x 3 of 19 Figure S2. 2-DE reference maps of secretomes; 150 μg proteins were loaded on an 11 cm IPG strip with a linear pH gradient PI 5–8 for IEF; 12% SDS-polyacrylamide gels were used for the second dimension. Proteins were stained with Flamingo fluorescent gel stain. Identified spots were assigned a number corresponding to that in their table. 2-DE maps from secretome of (A) TK173 control and (B) TGFβ1- treated ones. The 2-DE patterns revealed an alteration of secretome in stimulated TK173. Secretome patterns from TK173 treated with (C) ANG II and (D) PDGF. Cells 2020, 9, x 4 of 19 A Figure S3. Classification of the differentially expressed proteins upon ANG II, TGFβ1, or PDGF treatment in TK173. (A) Bar charts of the cellular component analyzed by STRAP biological function analysis in which the identified proteins from all treatments in both cell types are involved. -

The Structural Basis for Pyrophosphatase Catalysis
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Research Article 1491 The structural basis for pyrophosphatase catalysis Pirkko Heikinheimo1,2, Jukka Lehtonen1,2, Alexander Baykov3, Reijo Lahti2, Barry S Cooperman4* and Adrian Goldman1,2* Background: Soluble inorganic pyrophosphatase (PPase), an essential Addresses: 1Turku Centre for Biotechnology, PO enzyme central to phosphorus metabolism, catalyzes the hydrolysis of the phos- Box 123, FIN-20521 Turku, Finland, 2Department phoanhydride bond in inorganic pyrophosphate. Catalysis requires divalent of Biochemistry, University of Turku, FIN-20014 Turku, Finland, 3A.N. Belozersky Institute of metal ions which affect the apparent pKas of the essential general acid and Physico-Chemical Biology, Moscow State base on the enzyme, and the pKa of the substrate. Three to five metal ions are University, Moscow 119899, Russia and required for maximal activity, depending on pH and enzyme source. A detailed 4Department of Chemistry, University of understanding of catalysis would aid both in understanding the nature of biolog- Pennsylvania, Philadelphia, PA 19104, USA. ical mechanisms of phosphoryl transfer, and in understanding the role of diva- *Corresponding authors. lent cations. Without a high-resolution complex structure such a model has E-mail: [email protected] previously been unobtainable. E-mail: [email protected] Key words: mechanism, phosphoanhydride Results: We report the first two high-resolution structures of yeast PPase, at hydrolysis, phosphoryl transfer, pyrophosphatase, 2.2 and 2.0 Å resolution with R factors of around 17 %. One structure contains refinement, structure the two activating metal ions; the other, the product (MnPi)2 as well. -

Identification of Motifs in Cholera Toxin A1 Polypeptide That Are Required for Its Interaction with Human ADP-Ribosylation Factor 6 in a Bacterial Two-Hybrid System
Identification of motifs in cholera toxin A1 polypeptide that are required for its interaction with human ADP-ribosylation factor 6 in a bacterial two-hybrid system Michael G. Jobling and Randall K. Holmes* Department of Microbiology, University of Colorado Health Sciences Center, Denver, CO 80220 Edited by John J. Mekalanos, Harvard Medical School, Boston, MA, and approved October 20, 2000 (received for review September 14, 2000) The latent ADP-ribosyltransferase activity of cholera toxin (CT) that teins called ADP-ribosylation factors (ARFs; refs. 5 and 6). is activated after proteolytic nicking and reduction is associated ARFs are members of a highly conserved multigene family of with the CT A1 subunit (CTA1) polypeptide. This activity is stimu- small GTP-binding proteins, originally identified as activators of lated in vitro by interaction with eukaryotic proteins termed CT. They interact with several other proteins and are ubiqui- ADP-ribosylation factors (ARFs). We analyzed this interaction in a tously involved in membrane trafficking events (7). ARFs must modified bacterial two-hybrid system in which the T18 and T25 be in the GTP-bound form to be active. There are six mammalian fragments of the catalytic domain of Bordetella pertussis adenylate ARFs (five human) that fall into three classes: ARFs 1, 2, and cyclase were fused to CTA1 and human ARF6 polypeptides, respec- 3 (class I), ARFs 4 and 5 (class II), and ARF6 (class III). Class tively. Direct interaction between the CTA1 and ARF6 domains in I ARFs regulate the assembly of several types of vesicle coat these hybrid proteins reconstituted the adenylate cyclase activity complexes (8). -

In Vivo Mapping of a GPCR Interactome Using Knockin Mice
In vivo mapping of a GPCR interactome using knockin mice Jade Degrandmaisona,b,c,d,e,1, Khaled Abdallahb,c,d,1, Véronique Blaisb,c,d, Samuel Géniera,c,d, Marie-Pier Lalumièrea,c,d, Francis Bergeronb,c,d,e, Catherine M. Cahillf,g,h, Jim Boulterf,g,h, Christine L. Lavoieb,c,d,i, Jean-Luc Parenta,c,d,i,2, and Louis Gendronb,c,d,i,j,k,2 aDépartement de Médecine, Université de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; bDépartement de Pharmacologie–Physiologie, Université de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; cFaculté de Médecine et des Sciences de la Santé, Université de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; dCentre de Recherche du Centre Hospitalier Universitaire de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; eQuebec Network of Junior Pain Investigators, Sherbrooke, QC J1H 5N4, Canada; fDepartment of Psychiatry and Biobehavioral Sciences, University of California, Los Angeles, CA 90095; gSemel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, CA 90095; hShirley and Stefan Hatos Center for Neuropharmacology, University of California, Los Angeles, CA 90095; iInstitut de Pharmacologie de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; jDépartement d’Anesthésiologie, Université de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; and kQuebec Pain Research Network, Sherbrooke, QC J1H 5N4, Canada Edited by Brian K. Kobilka, Stanford University School of Medicine, Stanford, CA, and approved April 9, 2020 (received for review October 16, 2019) With over 30% of current medications targeting this family of attenuates pain hypersensitivities in several chronic pain models proteins, G-protein–coupled receptors (GPCRs) remain invaluable including neuropathic, inflammatory, diabetic, and cancer pain therapeutic targets. -

Lysophosphatidic Acid Activates Arf6 to Promote the Mesenchymal Malignancy of Renal Cancer
ARTICLE Received 6 Jun 2015 | Accepted 6 Jan 2016 | Published 8 Feb 2016 DOI: 10.1038/ncomms10656 OPEN Lysophosphatidic acid activates Arf6 to promote the mesenchymal malignancy of renal cancer Shigeru Hashimoto1,*, Shuji Mikami2,*,w, Hirokazu Sugino1, Ayumu Yoshikawa1, Ari Hashimoto1, Yasuhito Onodera1, Shotaro Furukawa1,3, Haruka Handa1, Tsukasa Oikawa1, Yasunori Okada4, Mototsugu Oya5 & Hisataka Sabe1 Acquisition of mesenchymal properties by cancer cells is critical for their malignant behaviour, but regulators of the mesenchymal molecular machinery and how it is activated remain elusive. Here we show that clear cell renal cell carcinomas (ccRCCs) frequently utilize the Arf6-based mesenchymal pathway to promote invasion and metastasis, similar to breast cancers. In breast cancer cells, ligand-activated receptor tyrosine kinases employ GEP100 to activate Arf6, which then recruits AMAP1; and AMAP1 then binds to the mesenchymal- specific protein EPB41L5, which promotes epithelial–mesenchymal transition and focal adhesion dynamics. In renal cancer cells, lysophosphatidic acid (LPA) activates Arf6 via its G-protein-coupled receptors, in which GTP-Ga12 binds to EFA6. The Arf6-based pathway may also contribute to drug resistance. Our results identify a specific mesenchymal molecular machinery of primary ccRCCs, which is triggered by a product of autotaxin and it is associated with poor outcome of patients. 1 Department of Molecular Biology, Graduate School of Medicine, Hokkaido University, Sapporo 060-8638, Japan. 2 Division of Diagnostic Pathology, Keio University Hospital, Tokyo 160-0016, Japan. 3 Department of Gastroenterological Surgery II, Graduate School of Medicine, Hokkaido University, Sapporo 060-8638, Japan. 4 Department of Pathology, Keio University School of Medicine, Tokyo 160-0016, Japan. -

Dysfunction Cytokine Release and Pancreatic Islet Graft Polypeptide-Induced Proinflammatory IL-1 Blockade Attenuates Islet Amylo
IL-1 Blockade Attenuates Islet Amyloid Polypeptide-Induced Proinflammatory Cytokine Release and Pancreatic Islet Graft Dysfunction This information is current as of September 23, 2021. Clara Westwell-Roper, Derek L. Dai, Galina Soukhatcheva, Kathryn J. Potter, Nico van Rooijen, Jan A. Ehses and C. Bruce Verchere J Immunol 2011; 187:2755-2765; Prepublished online 3 August 2011; Downloaded from doi: 10.4049/jimmunol.1002854 http://www.jimmunol.org/content/187/5/2755 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2011/08/04/jimmunol.100285 Material 4.DC1 References This article cites 57 articles, 21 of which you can access for free at: http://www.jimmunol.org/content/187/5/2755.full#ref-list-1 Why The JI? Submit online. by guest on September 23, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology IL-1 Blockade Attenuates Islet Amyloid Polypeptide-Induced Proinflammatory Cytokine Release and Pancreatic Islet Graft Dysfunction Clara Westwell-Roper,* Derek L. -

The Small Gtpase Arf6 Functions As a Membrane Tether in a Chemically-Defined Reconstitution System
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.12.380873; this version posted December 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The small GTPase Arf6 functions as a membrane tether in a chemically-defined reconstitution system Kana Fujibayashi and Joji Mima* Institute for Protein Research, Osaka University, Suita, Osaka 565-0871, Japan *Correspondence: [email protected] Abstract in all eukaryotic endomembrane systems and, in general, to function through cooperation with their specific interactors, Arf-family small GTPases are essential protein components termed Arf effectors (D’Souza-Schorey and Chavrier, 2006; for membrane trafficking in all eukaryotic endomembrane Donaldson and Jackson, 2011; Jackson and Bouvet, 2014; systems, particularly during the formation of membrane- Sztul et al., 2019). In particular, a large body of prior studies bound, coat protein complex-coated transport carriers. In on Arf-family small GTPases have established their pivotal addition to their roles in the transport carrier formation, a functions during the formation of membrane-bound, coat number of Arf-family GTPases have been reported to protein complex-coated transport carriers (e.g., secretory physically associate with coiled-coil tethering proteins and and endocytic transport vesicles) at the donor membranes multisubunit tethering complexes, which are responsible of subcellular compartments (D’Souza-Schorey and Chavrier, for membrane tethering, a process of the initial contact 2006; Sztul et al., 2019): Arf1 recruits the COPI (coat protein between transport carriers and their target subcellular complex I) subunits for initiating vesicle formation in the compartments. -

QKI Suppl Figs Tables Merged.Pdf
Supplemental Table 1 p<0.05 Human (Hs683) EntrezGene Name shQKI-1/shGFP shQKI-2/shGFP 0 0 26.63 19.00 0 0 11.75 12.75 0 CDNA clone IMAGE:5267346 8.96 4.77 LOC91316: Similar to bK246H3.1 (immunoglobulin lambda-like 91316 polypeptide 1, pre-B-cell specific) 7.94 3.85 RIPK2: receptor-interacting serine- 8767 threonine kinase 2 4.09 3.21 LOC728705: hypothetical protein 728705 LOC728705 3.86 2.44 0 Transcribed locus 3.79 3.95 CCDC57: coiled-coil domain containing 284001 57 3.62 4.73 C1orf101: chromosome 1 open reading 257044 frame 101 3.54 3.33 CDNA FLJ30490 fis, clone 0 BRAWH2000169 3.28 2.76 CHRNE: cholinergic receptor, nicotinic, 1145 epsilon 3.25 2.42 LOC728215: similar to transmembrane 728215 protein 28 3.25 2.86 83849 SYT15: Synaptotagmin XV 3.24 3.30 0 Transcribed locus 3.18 2.64 79838 TMC5: transmembrane channel-like 5 3.16 2.70 144568 A2ML1: alpha-2-macroglobulin-like 1 3.13 2.77 AGGF1: angiogenic factor with G patch 55109 and FHA domains 1 3.09 2.86 91703 ACY3: aspartoacylase (aminocyclase) 3 3.06 2.30 258010 SVIP: small VCP/p97-interacting protein 3.01 2.30 MSR1: macrophage scavenger receptor 4481 1 2.97 2.01 23250 ATP11A: ATPase, class VI, type 11A 2.96 1.71 LOC440934: Hypothetical gene 440934 supported by BC008048 2.89 2.27 0 Transcribed locus 2.87 3.04 Homo sapiens, clone IMAGE:4391558, 0 mRNA 2.85 3.38 C4orf38: chromosome 4 open reading 152641 frame 38 2.79 1.88 54756 IL17RD: interleukin 17 receptor D 2.77 1.98 0 CDNA clone IMAGE:5277449 2.75 2.25 OR51B2: olfactory receptor, family 51, 79345 subfamily B, member 2 2.75 1.89 0 -

ALLOSTERIC REGULATION of GS on AGONIST, ANTAGONIST and INVERSE AGONIST BINDING to the Β2ar by Gisselle A. Vélez Ruiz a Dissert
ALLOSTERIC REGULATION OF GS ON AGONIST, ANTAGONIST AND INVERSE AGONIST BINDING TO THE β2AR by Gisselle A. Vélez Ruiz A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Cellular and Molecular Biology) in the University of Michigan 2011 Doctoral Committee: Associate Professor Roger K. Sunahara, Chair Professor Richard R. Neubig Professor Michael D. Uhler Associate Professor Martin Myers Associate Professor John J.G. Tesmer To my family especially my mom and siblings, best friends: Grisel and Nelly, and to my boyfriend Osvaldo. Your constant love, support and eternal encouragement gave me the strength and motivation to accomplish my goals. ii ACKNOWLEDGEMENTS I would like to thank my mentor Roger Sunahara for all his support and endless enthusiasm. His support and patience allowed me to become a great researcher. I would also like to thank all the members of the Sunahara lab, past and present, especially Dr. Matthew Whorton and Dr. Adam Kuszak. Thank you for all your help and motivation especially when science was not on my side. Thank you for the great science but most importantly for the laughs; not matter how bad my day was they always found a way to make me laugh and I was always happy to be in lab even if I was not working. I would also like to thank Brian DeVree for all his help and endless conversations about GPCR theory and the implications of our research; a lot of those were documented here. I was extremely fortunate to have great collaborators that not only provided me with all the reagents I needed but also were key in making my project a success.