Mechanism of Guanosine Triphosphate Hydrolysis by the Visual Proteins Arl3-RP2: Free Energy Reaction Profiles Computed with Ab Initio Type QM/MM Potentials
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-Rab11 Antibody (ARG41900)
Product datasheet [email protected] ARG41900 Package: 100 μg anti-Rab11 antibody Store at: -20°C Summary Product Description Goat Polyclonal antibody recognizes Rab11 Tested Reactivity Hu, Ms, Rat, Dog, Mk Tested Application IHC-Fr, IHC-P, WB Host Goat Clonality Polyclonal Isotype IgG Target Name Rab11 Antigen Species Mouse Immunogen Purified recombinant peptides within aa. 110 to the C-terminus of Mouse Rab11a, Rab11b and Rab11c (Rab25). Conjugation Un-conjugated Alternate Names RAB11A: Rab-11; Ras-related protein Rab-11A; YL8 RAB11B: GTP-binding protein YPT3; H-YPT3; Ras-related protein Rab-11B RAB25: RAB11C; CATX-8; Ras-related protein Rab-25 Application Instructions Application table Application Dilution IHC-Fr 1:100 - 1:400 IHC-P 1:100 - 1:400 WB 1:250 - 1:2000 Application Note IHC-P: Antigen Retrieval: Heat mediation was recommended. * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control Hepa cell lysate Calculated Mw 24 kDa Observed Size ~ 26 kDa Properties Form Liquid Purification Affinity purification with immunogen. Buffer PBS, 0.05% Sodium azide and 20% Glycerol. Preservative 0.05% Sodium azide www.arigobio.com 1/3 Stabilizer 20% Glycerol Concentration 3 mg/ml Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. -

Retinal Dystrophy Panel Plus
ANALYSIS REPORT Patient: Test, Test 1; DoB: 1989-Apr-10; Order ID: 54324 SUMMARY DIAGNOSTIC FINDING Retinal Dystrophy Panel Plus PATIENT INFORMATION NAME DOB SSN AGE GENDER ORDER ID Test, test1 1989-Apr-10 10041989-xxxx 28 Male 54324 REFERRING HEALTHCARE PROFESSIONAL NAME HOSPITAL Anna Smith Test hospital SUMMARY OF CLINICAL HISTORY Patient is a 28-year-old male with VA 20/40 OD, 20/50 OS, constricted visual fields, non-detectable rod ffERG, and a family history consistent with X-linked retinitis pigmentosa. SEQUENCE ANALYSIS RESULTS GENE NOMENCLATURE INHERITANCE ZYGOSITY CLASSIFICATION RPGR c.2655_2656del, p.(Glu886Glyfs*192) X-linked HEM LIKELY PATHOGENIC DEL/DUP (CNV) ANALYSIS RESULTS Del/Dup (CNV) analysis did not detect any known disease-causing copy number variation or novel or rare deletion/duplication that was considered deleterious. CONCLUSION We classify the identifiedRPGR c.2655_2656del, p.(Glu886Glyfs*192) as likely pathogenic and the probable cause for the patient’s disease, considering the current evidence of the variant (established association between the gene and the patient’s phenotype, rarity in control populations, identification of the variant in an individual with the same phenotype, and mutation type: frameshift). However, additional information is still needed to confirm the pathogenicity of the variant, which could allow independent risk stratification based on this mutation. Genetic counseling and family member testing is recommended. Disease caused by RPGR mutations is inherited in an X-linked manner. We recommend carrier testing of the mother. If the mother is a carrier, then each offspring has a 50% chance of inheriting the mutation. Disease caused byRPGR mutations is inherited in an X-linked manner, and thus each daughter of an affected male will inherit the mutation, while sons will remain unaffected. -

Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum Brent Elliot Wells
The University of Maine DigitalCommons@UMaine Electronic Theses and Dissertations Fogler Library 2003 Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum Brent Elliot Wells Follow this and additional works at: http://digitalcommons.library.umaine.edu/etd Part of the Biochemistry Commons Recommended Citation Wells, Brent Elliot, "Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum" (2003). Electronic Theses and Dissertations. 301. http://digitalcommons.library.umaine.edu/etd/301 This Open-Access Thesis is brought to you for free and open access by DigitalCommons@UMaine. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of DigitalCommons@UMaine. IDENTIFICATION AND PARTIAL CHARACTERIZATION OF A FAMILY OF PUTATIVE PALMITOYLTRANSFERASES IN DICTYOSTELIUM DISCOIDEUM BY Brent Elliott Wells B.S. University of Utah, 1994 A THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science (in Biochemistry) The Graduate School The University of Maine December, 2003 Advisory Committee: Robert E. Gundersen, Associate Professor of Biochemistry, Advisor Keith W. Hutchison, Professor of Biochemistry Mary Rumpho-Kennedy, Professor of Biochemistry LIBRARY RIGHTS STATEMENT In presenting this thesis in partial fulfillment of the requirements for an advanced degree at The University of Maine, I agree that the Library shall make it freely available for inspection. I further agree that permission for "fair use" copying of this thesis for scholarly purposes may be granted by the Librarian. It is understood that any copying or publication of this thesis for financial gain shall not be allowed without my written permission. -

Protein Prenylation Reactions As Tools for Site-Specific Protein Labeling and Identification of Prenylation Substrates
Protein prenylation reactions as tools for site-specific protein labeling and identification of prenylation substrates Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) des Fachbereichs Chemie der Technischen Universität Dortmund Angefertigt am Max-Planck-Institut für molekulare Physiologie in Dortmund Vorgelegt von Dipl.-Chemikerin Thi Thanh Uyen Nguyen aus Jülich Dortmund, April 2009 Die vorliegende Arbeit wurde in der Zeit von Oktober 2005 bis April 2009 am Max-Planck- Institut für molekulare Physiologie in Dortmund unter der Anleitung von Prof. Dr. Roger S. Goody, Prof. Dr. Kirill Alexandrov und Prof. Dr. Herbert Waldmann durchgeführt. 1. Gutachter : Prof. Dr. R. S. Goody 2. Gutachter : Prof. Dr. H. Waldmann The results of this work were published in the following journals: “Exploiting the substrate tolerance of farnesyltransferase for site-selective protein derivatization” U.T.T. Nguyen, J. Cramer, J. Gomis, R. Reents, M. Gutierrez-Rodriguez, R.S. Goody, K. Alexandrov, H. Waldmann, Chembiochem 2007, 8 (4), 408-23. “Development of selective RabGGTase inhibitors and crystal structure of a RabGGTase- inhibitor complex” Z. Guo, Y.W. Wu, K.T. Tan, R.S. Bon, E. Guiu-Rozas, C. Delon, U.T.T. Nguyen, S. Wetzel,S. Arndt, R.S. Goody, W. Blankenfeldt, K. Alexandrov, H. Waldmann, Angew Chem Int Ed Engl 2008, 47 (20), 3747-50. “Analysis of the eukaryotic prenylome by isoprenoid affinity tagging” U.T.T. Nguyen, Z. Guo, C. Delon, Y.W. Wu, C. Deraeve, B. Franzel, R.S. Bon, W. Blankenfeldt, R.S. Goody, H. Waldmann, D. Wolters, K. Alexandrov, Nat Chem Biol 2009, 5 (4), 227-35. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Building the Vertebrate Codex Using the Gene Breaking Protein Trap Library
Genetics, Development and Cell Biology Publications Genetics, Development and Cell Biology 2020 Building the vertebrate codex using the gene breaking protein trap library Noriko Ichino Mayo Clinic Gaurav K. Varshney National Institutes of Health Ying Wang Iowa State University Hsin-kai Liao Iowa State University Maura McGrail Iowa State University, [email protected] See next page for additional authors Follow this and additional works at: https://lib.dr.iastate.edu/gdcb_las_pubs Part of the Molecular Genetics Commons, and the Research Methods in Life Sciences Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ gdcb_las_pubs/261. For information on how to cite this item, please visit http://lib.dr.iastate.edu/howtocite.html. This Article is brought to you for free and open access by the Genetics, Development and Cell Biology at Iowa State University Digital Repository. It has been accepted for inclusion in Genetics, Development and Cell Biology Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Building the vertebrate codex using the gene breaking protein trap library Abstract One key bottleneck in understanding the human genome is the relative under-characterization of 90% of protein coding regions. We report a collection of 1200 transgenic zebrafish strains made with the gene- break transposon (GBT) protein trap to simultaneously report and reversibly knockdown the tagged genes. Protein trap-associated mRFP expression shows previously undocumented expression of 35% and 90% of cloned genes at 2 and 4 days post-fertilization, respectively. Further, investigated alleles regularly show 99% gene-specific mRNA knockdown. -

RP2 Antibody / XRP2 (RQ4671)
RP2 Antibody / XRP2 (RQ4671) Catalog No. Formulation Size RQ4671 0.5mg/ml if reconstituted with 0.2ml sterile DI water 100 ug Bulk quote request Availability 1-3 business days Species Reactivity Human, Mouse, Rat Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit IgG Purity Antigen affinity purified Buffer Lyophilized from 1X PBS with 2% Trehalose and 0.025% sodium azide UniProt O75695 Applications Western blot : 0.5-1ug/ml Immunohistochemistry (FFPE) : 1-2ug/ml Immunofluorescence : 2-4ug/ml Flow cytometry : 1-3ug/million cells Direct ELISA : 0.1-0.5ug/ml (human recombinant protein) Limitations This RP2 antibody is available for research use only. Western blot testing of 1) human placenta, 2) human SGC-7901, 3) rat lung and 4) mouse lung lysate with RP2 antibody at 0.5ug/ml. Predicted molecular weight ~40 kDa. IHC staining of FFPE human placenta with RP2 antibody at 1ug/ml. HIER: boil tissue sections in pH6, 10mM citrate buffer, for 10-20 min followed by cooling at RT for 20 min. IHC staining of FFPE human placenta with RP2 antibody at 1ug/ml. HIER: boil tissue sections in pH6, 10mM citrate buffer, for 10-20 min followed by cooling at RT for 20 min. IHC staining of FFPE human lung cancer with RP2 antibody at 1ug/ml. HIER: boil tissue sections in pH6, 10mM citrate buffer, for 10-20 min followed by cooling at RT for 20 min. IHC staining of FFPE human breast cancer with RP2 antibody at 1ug/ml. HIER: boil tissue sections in pH6, 10mM citrate buffer, for 10-20 min followed by cooling at RT for 20 min. -

Pigmentosa in a Large Kindred
J Med Genet: first published as 10.1136/jmg.28.7.453 on 1 July 1991. Downloaded from I Med Genet 1991; 28: 453-457 453 Genetic localisation of the RP2 type of X linked retinitis pigmentosa in a large kindred A F Wright, SS Bhattacharya, M A Aldred, M Jay, A D Carothers, N S T Thomas, A C Bird, B Jay, H J Evans Abstract and usually loss of central vision before the fourth Genetic linkage and deletion studies have led to the decade. At least 14 to 15% of families and 15 to 28% of proposal that there are at least two loci on the X retinitis pigmentosa patients in the UK have the X chromosome which are responsible for X linked linked disorder' 2 so that the population prevalence is retinitis pigmentosa (XLRP). One locus (RP3) has in the region of 1 in 10 000 to 30 000. been closely defined by genetic linkage and deletion XLRP was first mapped to the proximal short arm analyses and localised to the region between the of the X chromosome by genetic linkage to DXS7, ornithine transcarbamylase (OTC) and chronic localised to Xpl l.-pll .3.3 Subsequent linkage granulomatous disease (CYBB) loci in Xp2l.1- studies have supported locations both proximal49 and pII.4. The other locus (RP2) has been assigned by distal'o'3 to the DXS7 locus. The situation was linkage analysis alone to region Xpll.4-pll.2, but clarified by a heterogeneity analysis of 62 XLRP its localisation is less weli defined. The results of a kindreds from nine centres, in which it was shown multipoint linkage analysis of a single large XLRP that the overall likelihood was 6-4x 109:1 in favour of kindred using eight informative loci provide further two loci versus a single XLRP locus. -

Inorganic Pyrophosphatase Is a Component of the Drosophila Nucleosome Remodeling Factor Complex
Downloaded from genesdev.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Inorganic pyrophosphatase is a component of the Drosophila nucleosome remodeling factor complex David A. Gdula, Raphael Sandaltzopoulos, Toshio Tsukiyama,1 Vincent Ossipow, and Carl Wu2 Laboratory of Molecular Cell Biology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892-4255 USA The Drosophila nucleosome remodeling factor (NURF) is a protein complex consisting of four polypeptides that facilitates the perturbation of chromatin structure in vitro in an ATP-dependent manner. The 140-kD NURF subunit, imitation switch (ISWI), is related to the SWI2/SNF2 ATPase. Another subunit, NURF-55, is a 55-kD WD repeat protein homologous to the human retinoblastoma-associated protein RbAp48. Here, we report the cloning and characterization of the smallest (38 kD) component of NURF. NURF-38 is strikingly homologous to known inorganic pyrophosphatases. Both recombinant NURF-38 alone and the purified NURF complex are shown to have inorganic pyrophosphatase activity. Inhibition of the pyrophosphatase activity of NURF with sodium fluoride has no significant effect on chromatin remodeling, indicating that these two activities may be biochemically uncoupled. Our results suggest that NURF-38 may serve a structural or regulatory role in the complex. Alternatively, because accumulation of unhydrolyzed pyrophosphate during nucleotide incorporation inhibits polymerization, NURF may also have been adapted to deliver pyrophosphatase -

A Chemical Proteomic Approach to Investigate Rab Prenylation in Living Systems
A chemical proteomic approach to investigate Rab prenylation in living systems By Alexandra Fay Helen Berry A thesis submitted to Imperial College London in candidature for the degree of Doctor of Philosophy of Imperial College. Department of Chemistry Imperial College London Exhibition Road London SW7 2AZ August 2012 Declaration of Originality I, Alexandra Fay Helen Berry, hereby declare that this thesis, and all the work presented in it, is my own and that it has been generated by me as the result of my own original research, unless otherwise stated. 2 Abstract Protein prenylation is an important post-translational modification that occurs in all eukaryotes; defects in the prenylation machinery can lead to toxicity or pathogenesis. Prenylation is the modification of a protein with a farnesyl or geranylgeranyl isoprenoid, and it facilitates protein- membrane and protein-protein interactions. Proteins of the Ras superfamily of small GTPases are almost all prenylated and of these the Rab family of proteins forms the largest group. Rab proteins are geranylgeranylated with up to two geranylgeranyl groups by the enzyme Rab geranylgeranyltransferase (RGGT). Prenylation of Rabs allows them to locate to the correct intracellular membranes and carry out their roles in vesicle trafficking. Traditional methods for probing prenylation involve the use of tritiated geranylgeranyl pyrophosphate which is hazardous, has lengthy detection times, and is insufficiently sensitive. The work described in this thesis developed systems for labelling Rabs and other geranylgeranylated proteins using a technique known as tagging-by-substrate, enabling rapid analysis of defective Rab prenylation in cells and tissues. An azide analogue of the geranylgeranyl pyrophosphate substrate of RGGT (AzGGpp) was applied for in vitro prenylation of Rabs by recombinant enzyme. -
Type of the Paper (Article
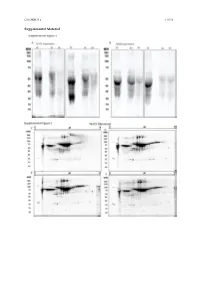
Cells 2020, 9, x 1 of 19 Supplemental Material Cells 2020, 9, x 2 of 19 Figure S1. Secretome enrichment: protocol optimization. 1D SDS-PAGE documentation of washing steps: Culture medium was substituted with FCS-free medium, which was changed every 2 h. The supernatants were then collected, and the proteins isolated and separated in 1D SDS-PAGE ((A) TK173 and (B) TK188). Proteins were stained with Flamingo fluorescent gel stain. Two-dimensional pattern of the proteins isolated from supernatant of TK173, (C) 2 h, (D) 4 h, (E) 6 h, and (F) 8 h after changing to FCS-free medium. (G) Cell secretome collected 24 h after elimination of the contaminating FCS-proteins with different washing steps. Proteins were stained with Flamingo fluorescent gel stain. Cells 2020, 9, x 3 of 19 Figure S2. 2-DE reference maps of secretomes; 150 μg proteins were loaded on an 11 cm IPG strip with a linear pH gradient PI 5–8 for IEF; 12% SDS-polyacrylamide gels were used for the second dimension. Proteins were stained with Flamingo fluorescent gel stain. Identified spots were assigned a number corresponding to that in their table. 2-DE maps from secretome of (A) TK173 control and (B) TGFβ1- treated ones. The 2-DE patterns revealed an alteration of secretome in stimulated TK173. Secretome patterns from TK173 treated with (C) ANG II and (D) PDGF. Cells 2020, 9, x 4 of 19 A Figure S3. Classification of the differentially expressed proteins upon ANG II, TGFβ1, or PDGF treatment in TK173. (A) Bar charts of the cellular component analyzed by STRAP biological function analysis in which the identified proteins from all treatments in both cell types are involved. -

The Structural Basis for Pyrophosphatase Catalysis
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Research Article 1491 The structural basis for pyrophosphatase catalysis Pirkko Heikinheimo1,2, Jukka Lehtonen1,2, Alexander Baykov3, Reijo Lahti2, Barry S Cooperman4* and Adrian Goldman1,2* Background: Soluble inorganic pyrophosphatase (PPase), an essential Addresses: 1Turku Centre for Biotechnology, PO enzyme central to phosphorus metabolism, catalyzes the hydrolysis of the phos- Box 123, FIN-20521 Turku, Finland, 2Department phoanhydride bond in inorganic pyrophosphate. Catalysis requires divalent of Biochemistry, University of Turku, FIN-20014 Turku, Finland, 3A.N. Belozersky Institute of metal ions which affect the apparent pKas of the essential general acid and Physico-Chemical Biology, Moscow State base on the enzyme, and the pKa of the substrate. Three to five metal ions are University, Moscow 119899, Russia and required for maximal activity, depending on pH and enzyme source. A detailed 4Department of Chemistry, University of understanding of catalysis would aid both in understanding the nature of biolog- Pennsylvania, Philadelphia, PA 19104, USA. ical mechanisms of phosphoryl transfer, and in understanding the role of diva- *Corresponding authors. lent cations. Without a high-resolution complex structure such a model has E-mail: [email protected] previously been unobtainable. E-mail: [email protected] Key words: mechanism, phosphoanhydride Results: We report the first two high-resolution structures of yeast PPase, at hydrolysis, phosphoryl transfer, pyrophosphatase, 2.2 and 2.0 Å resolution with R factors of around 17 %. One structure contains refinement, structure the two activating metal ions; the other, the product (MnPi)2 as well.