Supplemental Methods Cell Culture, Cell Lines and Transgenic Mice. for Construction of the MMTV/Cyclin D1 Vector the Cdna Encodi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
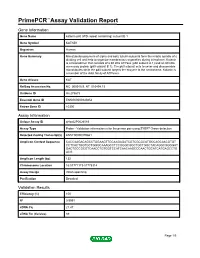
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name katanin p80 (WD repeat containing) subunit B 1 Gene Symbol KATNB1 Organism Human Gene Summary Microtubules polymers of alpha and beta tubulin subunits form the mitotic spindle of a dividing cell and help to organize membranous organelles during interphase. Katanin is a heterodimer that consists of a 60 kDa ATPase (p60 subunit A 1) and an 80 kDa accessory protein (p80 subunit B 1). The p60 subunit acts to sever and disassemble microtubules while the p80 subunit targets the enzyme to the centrosome. Katanin is a member of the AAA family of ATPases. Gene Aliases KAT RefSeq Accession No. NC_000016.9, NT_010498.15 UniGene ID Hs.275675 Ensembl Gene ID ENSG00000140854 Entrez Gene ID 10300 Assay Information Unique Assay ID qHsaCIP0026589 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000379661 Amplicon Context Sequence CACCAAGACAGCCTGGAAGTTGCAAGAGATCGTCGCGCATGCCAGCAACGTGT CCTCACTGGTGCTGGGCAAAGCCTCCGGGCGGCTGCTGGCTACAGGCGGGGAT GACTGCCGCGTCAACCTGTGGTCCATCAACAAGCCCAACTGCATCATGAGCCTG ACG Amplicon Length (bp) 132 Chromosome Location 16:57771173-57778314 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9991 cDNA Cq 21.47 cDNA Tm (Celsius) 89 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 35.43 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report KATNB1, Human Amplification -

Polyclonal Antibody to APC11 / ANAPC11 - Serum
OriGene Technologies, Inc. OriGene Technologies GmbH 9620 Medical Center Drive, Ste 200 Schillerstr. 5 Rockville, MD 20850 32052 Herford UNITED STATES GERMANY Phone: +1-888-267-4436 Phone: +49-5221-34606-0 Fax: +1-301-340-8606 Fax: +49-5221-34606-11 [email protected] [email protected] R1503 Polyclonal Antibody to APC11 / ANAPC11 - Serum Alternate names: Anaphase-promoting complex subunit 11, Cyclosome subunit 11, HSPC214, Hepatocellular carcinoma-associated RING finger protein Quantity: 0.1 ml Concentration: 85 mg/ml (by Refractometry) Background: APC11 is also known as Anaphase promoting complex subunit 11, APC11, Cyclosome subunit 11, Hepatocellular carcinoma associated RING finger protein, and HSPC214. APC11 is a component of the anaphase promoting complex/cyclosome (APC/C), a cell cycle-regulated E3 ubiquitin ligase that controls progression through mitosis and the G1 phase of the cell cycle. APC11 may function to recruit the E2 ubiquitin-conjugating enzymes to the complex. APC11 interacts with the cullin domain of ANAPC2 and also interacts with UBE2D2. APC11 shows both a cytoplasmic and nuclear localization. APC11 is expressed at high levels in skeletal muscle and heart; in moderate levels in brain, kidney, and liver; and at low levels in colon, thymus, spleen, small intestine, placenta, lung and peripheral blood leukocyte. APC11 is a member of the RING-type zinc finger family and is auto-ubiquitinylated. Uniprot ID: Q9NYG5 NCBI: NP_001002244.1 GeneID: 51529 Host: Rabbit Immunogen: This APC11 antibody was prepared from whole rabbit serum produced by repeated immunizations with a synthetic peptide corresponding to amino acids 76-84 of Human APC11 (C-terminal) coupled to KLH. -

KIAA0556 Is a Novel Ciliary Basal Body Component Mutated in Joubert Syndrome Anna A
Sanders et al. Genome Biology (2015) 16:293 DOI 10.1186/s13059-015-0858-z RESEARCH Open Access KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome Anna A. W. M. Sanders1†, Erik de Vrieze2,3†, Anas M. Alazami4†, Fatema Alzahrani4, Erik B. Malarkey5, Nasrin Sorusch6, Lars Tebbe6, Stefanie Kuhns1, Teunis J. P. van Dam7, Amal Alhashem8, Brahim Tabarki8, Qianhao Lu9,10, Nils J. Lambacher1, Julie E. Kennedy1, Rachel V. Bowie1, Lisette Hetterschijt2,3, Sylvia van Beersum3,11, Jeroen van Reeuwijk3,11, Karsten Boldt12, Hannie Kremer2,3,11, Robert A. Kesterson13, Dorota Monies4, Mohamed Abouelhoda4, Ronald Roepman3,11, Martijn H. Huynen7, Marius Ueffing12, Rob B. Russell9,10, Uwe Wolfrum6, Bradley K. Yoder5, Erwin van Wijk2,3*, Fowzan S. Alkuraya4,14* and Oliver E. Blacque1* Abstract Background: Joubert syndrome (JBTS) and related disorders are defined by cerebellar malformation (molar tooth sign), together with neurological symptoms of variable expressivity. The ciliary basis of Joubert syndrome related disorders frequently extends the phenotype to tissues such as the eye, kidney, skeleton and craniofacial structures. Results: Using autozygome and exome analyses, we identified a null mutation in KIAA0556 in a multiplex consanguineous family with hallmark features of mild Joubert syndrome. Patient-derived fibroblasts displayed reduced ciliogenesis potential and abnormally elongated cilia. Investigation of disease pathophysiology revealed that Kiaa0556-/- null mice possess a Joubert syndrome-associated brain-restricted phenotype. Functional studies in Caenorhabditis elegans nematodes and cultured human cells support a conserved ciliary role for KIAA0556 linked to microtubule regulation. First, nematode KIAA0556 is expressed almost exclusively in ciliated cells, and the worm and human KIAA0556 proteins are enriched at the ciliary base. -

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/) -

Katanin-P80 Gene Promoter Characterization and Regulation Via Elk1
Katanin-p80 Gene Promoter Characterization and Regulation via Elk1 Ece Selc¸uk., Koray Kırımtay., Derya Canbaz, Gu¨ her Is¸ık Cesur, Sirin Korulu, Arzu Karabay* Department of Molecular Biology and Genetics, Istanbul Technical University, Istanbul, Turkey Abstract Katanin is an ATPase family member protein that participates in microtubule severing. It has heterodimeric structure consisting of 60 kDa (katanin-p60) and 80 kDa (katanin-p80) subunits encoded by KATNA1 and KATNB1 genes, respectively. Katanin-p60 has the enzymatic activity for microtubule severing, whereas katanin-p80 consists of multiple domains with different functions such as targeting katanin-p60 to the centrosome, augmenting microtubule severing by katanin-p60, and even suppressing microtubule severing. Despite the various important functions of katanin-p80, its transcriptional regulation has not been studied yet. Elk1 transcription factor has been shown to interact with microtubules and regulate the transcription of another microtubule severing protein, spastin. In spite of katanin’s importance, and structural and functional similarities to spastin, there is no study on the transcriptional regulation of katanin yet. In this study, we aimed to characterize KATNB1 promoter and analyze the effects of Elk1 on katanin-p80 expression. We identified a 518- bp TATA-less promoter including a critical CpG island and GC boxes as an optimal promoter, and sequential deletion of CpG island and the GC elements gradually decreased the KATNB1 promoter activity. In addition, we showed Elk1 binding on the KATNB1 promoter by EMSA. We found that Elk1 activated KATNB1 promoter, and increased both mRNA and protein levels of katanin- p80 in SH-SY5Y cells. On the other hand, KCl treatment increasing SUMOylation decreased KATNB1 promoter activity. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

APC/C Is Essential for Hematopoiesis and Impaired in Aplastic Anemia
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 38), pp: 63360-63369 Research Paper APC/C is essential for hematopoiesis and impaired in aplastic anemia Jia Wang1,*, Min-Zhi Yin2,*, Ke-Wen Zhao1,*, Fang Ke1,*, Wen-Jie Jin3, Xiao-Lin Guo1, Tian-Hui Liu1, Xiao-Ye Liu1, Hao Gu1, Xiao-Min Yu1, Zhen Li1, Li-Li Mu1, Deng-Li Hong1, Jing Chen4 and Guo-Qiang Chen1 1Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Department of Pathophysiology, Shanghai Jiao Tong University School of Medicine (SJTU-SM), Shanghai, 200025, China 2Department of Pathology, Shanghai Children’s Medical Center, SJTU-SM, Shanghai, 200025, China 3Department of Orthopaedics, Shanghai Ninth People's Hospital, SJTU-SM, Shanghai, 200025, China 4Key Laboratory of Pediatric Hematology and Oncology Ministry of Health, Department of Hematology and Oncology, Shanghai Children’s Medical Center, SJTU-SM, Shanghai, 200127, China *These authors have contributed equally to this work Correspondence to: Guo-Qiang Chen, email: [email protected] Jing Chen, email: [email protected] Deng-Li Hong, email: [email protected] Keywords: anaphase promoting complex/cyclosome (APC/C), Anapc2, hematopoietic stem and progenitor cells (HSPCs), dormant HPSCs, aplastic anemia Received: November 22, 2016 Accepted: June 02, 2017 Published: June 28, 2017 Copyright: Wang et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Anaphase promoting complex/cyclosome (APC/C) is essential for cell cycle progression. -

The Genome of Schmidtea Mediterranea and the Evolution Of
OPEN ArtICLE doi:10.1038/nature25473 The genome of Schmidtea mediterranea and the evolution of core cellular mechanisms Markus Alexander Grohme1*, Siegfried Schloissnig2*, Andrei Rozanski1, Martin Pippel2, George Robert Young3, Sylke Winkler1, Holger Brandl1, Ian Henry1, Andreas Dahl4, Sean Powell2, Michael Hiller1,5, Eugene Myers1 & Jochen Christian Rink1 The planarian Schmidtea mediterranea is an important model for stem cell research and regeneration, but adequate genome resources for this species have been lacking. Here we report a highly contiguous genome assembly of S. mediterranea, using long-read sequencing and a de novo assembler (MARVEL) enhanced for low-complexity reads. The S. mediterranea genome is highly polymorphic and repetitive, and harbours a novel class of giant retroelements. Furthermore, the genome assembly lacks a number of highly conserved genes, including critical components of the mitotic spindle assembly checkpoint, but planarians maintain checkpoint function. Our genome assembly provides a key model system resource that will be useful for studying regeneration and the evolutionary plasticity of core cell biological mechanisms. Rapid regeneration from tiny pieces of tissue makes planarians a prime De novo long read assembly of the planarian genome model system for regeneration. Abundant adult pluripotent stem cells, In preparation for genome sequencing, we inbred the sexual strain termed neoblasts, power regeneration and the continuous turnover of S. mediterranea (Fig. 1a) for more than 17 successive sib- mating of all cell types1–3, and transplantation of a single neoblast can rescue generations in the hope of decreasing heterozygosity. We also developed a lethally irradiated animal4. Planarians therefore also constitute a a new DNA isolation protocol that meets the purity and high molecular prime model system for stem cell pluripotency and its evolutionary weight requirements of PacBio long-read sequencing12 (Extended Data underpinnings5. -

Table S2.Up Or Down Regulated Genes in Tcof1 Knockdown Neuroblastoma N1E-115 Cells Involved in Differentbiological Process Anal
Table S2.Up or down regulated genes in Tcof1 knockdown neuroblastoma N1E-115 cells involved in differentbiological process analysed by DAVID database Pop Pop Fold Term PValue Genes Bonferroni Benjamini FDR Hits Total Enrichment GO:0044257~cellular protein catabolic 2.77E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 537 13588 1.944851 8.64E-07 8.64E-07 5.02E-07 process ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, USP17L5, FBXO11, RAD23B, NEDD8, UBE2V2, RFFL, CDC GO:0051603~proteolysis involved in 4.52E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 534 13588 1.93519 1.41E-06 7.04E-07 8.18E-07 cellular protein catabolic process ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, USP17L5, FBXO11, RAD23B, NEDD8, UBE2V2, RFFL, CDC GO:0044265~cellular macromolecule 6.09E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 609 13588 1.859332 1.90E-06 6.32E-07 1.10E-06 catabolic process ISG15, RBM8A, ATG7, LOC100046898, PSENEN, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, XRN2, USP17L5, FBXO11, RAD23B, UBE2V2, NED GO:0030163~protein catabolic process 1.81E-09 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 556 13588 1.87839 5.64E-06 1.41E-06 3.27E-06 ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, -

A Transcriptional Signature of Postmitotic Maintenance in Neural Tissues
Neurobiology of Aging 74 (2019) 147e160 Contents lists available at ScienceDirect Neurobiology of Aging journal homepage: www.elsevier.com/locate/neuaging Postmitotic cell longevityeassociated genes: a transcriptional signature of postmitotic maintenance in neural tissues Atahualpa Castillo-Morales a,b, Jimena Monzón-Sandoval a,b, Araxi O. Urrutia b,c,*, Humberto Gutiérrez a,** a School of Life Sciences, University of Lincoln, Lincoln, UK b Milner Centre for Evolution, Department of Biology and Biochemistry, University of Bath, Bath, UK c Instituto de Ecología, Universidad Nacional Autónoma de México, Ciudad de México, Mexico article info abstract Article history: Different cell types have different postmitotic maintenance requirements. Nerve cells, however, are Received 11 April 2018 unique in this respect as they need to survive and preserve their functional complexity for the entire Received in revised form 3 October 2018 lifetime of the organism, and failure at any level of their supporting mechanisms leads to a wide range of Accepted 11 October 2018 neurodegenerative conditions. Whether these differences across tissues arise from the activation of Available online 19 October 2018 distinct cell typeespecific maintenance mechanisms or the differential activation of a common molecular repertoire is not known. To identify the transcriptional signature of postmitotic cellular longevity (PMCL), Keywords: we compared whole-genome transcriptome data from human tissues ranging in longevity from 120 days Neural maintenance Cell longevity to over 70 years and found a set of 81 genes whose expression levels are closely associated with Transcriptional signature increased cell longevity. Using expression data from 10 independent sources, we found that these genes Functional genomics are more highly coexpressed in longer-living tissues and are enriched in specific biological processes and transcription factor targets compared with randomly selected gene samples. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

2Q13 Microdeletions
2q13 microdeletions rarechromo.org 2q13 microdeletions A 2q13 microdeletion is a rare genetic condition caused by a small piece of missing genetic material from one of the body’s chromosomes - chromosome 2. Deletions can vary in size but those that are too small to be visible under the microscope using standard techniques are called microdeletions. For typical and healthy development, chromosomes should contain the expected amount of genetic material. Like most other chromosome disorders, having a missing piece of chromosome 2 may affect the development and intellectual abilities of a child. The outcome of having a 2q13 microdeletion is very variable and depends on a number of factors including what and how much genetic material is missing. Background on chromosomes Our bodies are made up of different types of cells, almost all of which contain the same chromosomes. Each chromosome consists of DNA that carries the code for hundreds to thousands of genes. Genes can be thought of as individual instruction booklets (or recipes) that contain all the genetic information that tells the body how to develop, grow and function. Chromosomes (and hence genes) usually come in pairs with one member of each chromosome pair being inherited from each parent. Most cells of the human body have a total of 46 (23 pairs of) chromosomes. The egg and the sperm cells, however, have 23 unpaired chromosomes, so that when the egg and sperm join together at conception, the chromosomes pair up to make a total of 46. Of these 46 chromosomes, 44 are grouped in 22 pairs, numbered 1 to 22.