Retroviral Vector Integration Deregulates Gene Expression but Has No Consequence on the Biology and Function of Transplanted T Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
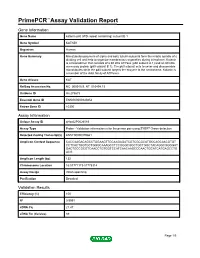
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name katanin p80 (WD repeat containing) subunit B 1 Gene Symbol KATNB1 Organism Human Gene Summary Microtubules polymers of alpha and beta tubulin subunits form the mitotic spindle of a dividing cell and help to organize membranous organelles during interphase. Katanin is a heterodimer that consists of a 60 kDa ATPase (p60 subunit A 1) and an 80 kDa accessory protein (p80 subunit B 1). The p60 subunit acts to sever and disassemble microtubules while the p80 subunit targets the enzyme to the centrosome. Katanin is a member of the AAA family of ATPases. Gene Aliases KAT RefSeq Accession No. NC_000016.9, NT_010498.15 UniGene ID Hs.275675 Ensembl Gene ID ENSG00000140854 Entrez Gene ID 10300 Assay Information Unique Assay ID qHsaCIP0026589 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000379661 Amplicon Context Sequence CACCAAGACAGCCTGGAAGTTGCAAGAGATCGTCGCGCATGCCAGCAACGTGT CCTCACTGGTGCTGGGCAAAGCCTCCGGGCGGCTGCTGGCTACAGGCGGGGAT GACTGCCGCGTCAACCTGTGGTCCATCAACAAGCCCAACTGCATCATGAGCCTG ACG Amplicon Length (bp) 132 Chromosome Location 16:57771173-57778314 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9991 cDNA Cq 21.47 cDNA Tm (Celsius) 89 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 35.43 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report KATNB1, Human Amplification -

KIAA0556 Is a Novel Ciliary Basal Body Component Mutated in Joubert Syndrome Anna A
Sanders et al. Genome Biology (2015) 16:293 DOI 10.1186/s13059-015-0858-z RESEARCH Open Access KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome Anna A. W. M. Sanders1†, Erik de Vrieze2,3†, Anas M. Alazami4†, Fatema Alzahrani4, Erik B. Malarkey5, Nasrin Sorusch6, Lars Tebbe6, Stefanie Kuhns1, Teunis J. P. van Dam7, Amal Alhashem8, Brahim Tabarki8, Qianhao Lu9,10, Nils J. Lambacher1, Julie E. Kennedy1, Rachel V. Bowie1, Lisette Hetterschijt2,3, Sylvia van Beersum3,11, Jeroen van Reeuwijk3,11, Karsten Boldt12, Hannie Kremer2,3,11, Robert A. Kesterson13, Dorota Monies4, Mohamed Abouelhoda4, Ronald Roepman3,11, Martijn H. Huynen7, Marius Ueffing12, Rob B. Russell9,10, Uwe Wolfrum6, Bradley K. Yoder5, Erwin van Wijk2,3*, Fowzan S. Alkuraya4,14* and Oliver E. Blacque1* Abstract Background: Joubert syndrome (JBTS) and related disorders are defined by cerebellar malformation (molar tooth sign), together with neurological symptoms of variable expressivity. The ciliary basis of Joubert syndrome related disorders frequently extends the phenotype to tissues such as the eye, kidney, skeleton and craniofacial structures. Results: Using autozygome and exome analyses, we identified a null mutation in KIAA0556 in a multiplex consanguineous family with hallmark features of mild Joubert syndrome. Patient-derived fibroblasts displayed reduced ciliogenesis potential and abnormally elongated cilia. Investigation of disease pathophysiology revealed that Kiaa0556-/- null mice possess a Joubert syndrome-associated brain-restricted phenotype. Functional studies in Caenorhabditis elegans nematodes and cultured human cells support a conserved ciliary role for KIAA0556 linked to microtubule regulation. First, nematode KIAA0556 is expressed almost exclusively in ciliated cells, and the worm and human KIAA0556 proteins are enriched at the ciliary base. -

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -

Katanin-P80 Gene Promoter Characterization and Regulation Via Elk1
Katanin-p80 Gene Promoter Characterization and Regulation via Elk1 Ece Selc¸uk., Koray Kırımtay., Derya Canbaz, Gu¨ her Is¸ık Cesur, Sirin Korulu, Arzu Karabay* Department of Molecular Biology and Genetics, Istanbul Technical University, Istanbul, Turkey Abstract Katanin is an ATPase family member protein that participates in microtubule severing. It has heterodimeric structure consisting of 60 kDa (katanin-p60) and 80 kDa (katanin-p80) subunits encoded by KATNA1 and KATNB1 genes, respectively. Katanin-p60 has the enzymatic activity for microtubule severing, whereas katanin-p80 consists of multiple domains with different functions such as targeting katanin-p60 to the centrosome, augmenting microtubule severing by katanin-p60, and even suppressing microtubule severing. Despite the various important functions of katanin-p80, its transcriptional regulation has not been studied yet. Elk1 transcription factor has been shown to interact with microtubules and regulate the transcription of another microtubule severing protein, spastin. In spite of katanin’s importance, and structural and functional similarities to spastin, there is no study on the transcriptional regulation of katanin yet. In this study, we aimed to characterize KATNB1 promoter and analyze the effects of Elk1 on katanin-p80 expression. We identified a 518- bp TATA-less promoter including a critical CpG island and GC boxes as an optimal promoter, and sequential deletion of CpG island and the GC elements gradually decreased the KATNB1 promoter activity. In addition, we showed Elk1 binding on the KATNB1 promoter by EMSA. We found that Elk1 activated KATNB1 promoter, and increased both mRNA and protein levels of katanin- p80 in SH-SY5Y cells. On the other hand, KCl treatment increasing SUMOylation decreased KATNB1 promoter activity. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Supplemental Material 1
SUP. FIGURE S1 DAPI NFAT Merge DMSO Ac5SGlcNAc Figure S1. Inhibition of OGT does not prevent nuclear translocation of NFAT. Jurkat cells !"#$%&'()*+(!!,-.'/012"#..(3'4056'7(+('"+(#"(3'7,"8'9:';<'5=29>/%=45='?+'@<>A'B?+'CD' 8+!E''F(%%!'7(+('"8(-'*%#"(3'?-'#-",2F@GHF@ID2=?#"(3'=?J(+!%,*!'B?+'G:'K,-!E''5B"(+'B,)#",?-L' =(%%!'7(+('#-#%&M(3'$&'=?-B?=#%'K,=+?!=?*&E SUP. FIGURE S2 Labeled Unlabeled Labeled Unlabeled 1h 18h Az PEG 1h 19h Az PEG !"#$%"#&'( + + + + !"#$%"#&'( + + + + 110 EWSR1 160 *34+&5 80 &,/ 160 )*+-2$ 160 110 )*+,& 80 60 &,/ 160 110 80 0"1"- RUNX1 110 80 60 60 50 160 SP1 160 110 ELF1 110 80 160 NUP98 110 80 )*+&-. &,/ Figure S2. PEG mass tags allow visualization of OGlcNAc stoichiometry. 5K PEG mass tags were affixed to OGlcNAc groups on proteins from control or activated T cells via enzy matic labeling with azide and copperfree click chemistry. Proteins were then analyzed for shifts in electrophoretic mobility by immunoblot. For the unlabeled control samples, either the azide (Az) or PEG (PEG) reagent was omitted during the labeling procedure. Note that HCFC1 appears as multiple bands because the protein is expressed as a single polypeptide that under goes proteolytic processing. UBAP2L appears as two bands in the unlabeled control samples due to alternative splicing. Table S1. Details of 133 higher confidence and 81 lower confidence O-GlcNAc glycoproteins1. Confidence Uniprot ID Symbol Specificity Chi Higher P55265 ADAR 100% 9.11E-04 Higher Q09666 AHNAK 92% 2.36E-16 Higher Q8IWZ3 ANKHD1 100% 1.19E-03 Higher -

Ultraconserved Elements Are Associated with Homeostatic Control of Splicing Regulators by Alternative Splicing and Nonsense-Mediated Decay
Downloaded from genesdev.cshlp.org on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay Julie Z. Ni,1 Leslie Grate,1 John Paul Donohue,1 Christine Preston,2 Naomi Nobida,2 Georgeann O’Brien,2 Lily Shiue,1 Tyson A. Clark,3 John E. Blume,3 and Manuel Ares Jr.1,2,4 1Center for Molecular Biology of RNA and Department of Molecular, Cell, and Developmental Biology, University of California at Santa Cruz, Santa Cruz, California 95064, USA; 2Hughes Undergraduate Research Laboratory, University of California at Santa Cruz, Santa Cruz, California 95064, USA; 3Affymetrix, Inc., Santa Clara, California 95051, USA Many alternative splicing events create RNAs with premature stop codons, suggesting that alternative splicing coupled with nonsense-mediated decay (AS-NMD) may regulate gene expression post-transcriptionally. We tested this idea in mice by blocking NMD and measuring changes in isoform representation using splicing-sensitive microarrays. We found a striking class of highly conserved stop codon-containing exons whose inclusion renders the transcript sensitive to NMD. A genomic search for additional examples identified >50 such exons in genes with a variety of functions. These exons are unusually frequent in genes that encode splicing activators and are unexpectedly enriched in the so-called “ultraconserved” elements in the mammalian lineage. Further analysis show that NMD of mRNAs for splicing activators such as SR proteins is triggered by splicing activation events, whereas NMD of the mRNAs for negatively acting hnRNP proteins is triggered by splicing repression, a polarity consistent with widespread homeostatic control of splicing regulator gene expression. -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase.