Characterization and Engineering of Epimerases for the Production of Rare Sugars
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Atpase Domain Common to Prokaryotic Cell Cycle Proteins
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 7290-7294, August 1992 Biochemistry An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp7O heat shock proteins (structural comparison/property pattern/remote homology) PEER BORK, CHRIS SANDER, AND ALFONSO VALENCIA European Molecular Biology Laboratory, D-6900 Heidelberg, Federal Republic of Germany Communicated by Russell F. Doolittle, March 6, 1992 ABSTRACT The functionally diverse actin, hexokinase, and hsp7O protein families have in common an ATPase domain of known three-dimensional structure. Optimal superposition ofthe three structures and alignment ofmany sequences in each of the three families has revealed a set of common conserved residues, distributed in five sequence motifs, which are in- volved in ATP binding and in a putative interdomain hinge. From the multiple sequence aliment in these motifs a pattern of amino acid properties required at each position is defined. The discriminatory power of the pattern is in part due to the use of several known three-dimensional structures and many sequences and in part to the "property" method ofgeneralizing from observed amino acid frequencies to amino acid fitness at each sequence position. A sequence data base search with the pattern significantly matches sugar kinases, such as fuco-, glucono-, xylulo-, ribulo-, and glycerokinase, as well as the prokaryotic cell cycle proteins MreB, FtsA, and StbA. These are predicted to have subdomains with the same tertiary structure as the ATPase subdomains Ia and Ha of hexokinase, actin, and Hsc7O, a very similar ATP binding pocket, and the capacity for interdomain hinge motion accompanying func- tional state changes. -

Comparison of Strand-Specific Transcriptomes Of
Landstorfer et al. BMC Genomics 2014, 15:353 http://www.biomedcentral.com/1471-2164/15/353 RESEARCH ARTICLE Open Access Comparison of strand-specific transcriptomes of enterohemorrhagic Escherichia coli O157:H7 EDL933 (EHEC) under eleven different environmental conditions including radish sprouts and cattle feces Richard Landstorfer1, Svenja Simon2, Steffen Schober3, Daniel Keim2, Siegfried Scherer1 and Klaus Neuhaus1* Abstract Background: Multiple infection sources for enterohemorrhagic Escherichia coli O157:H7 (EHEC) are known, including animal products, fruit and vegetables. The ecology of this pathogen outside its human host is largely unknown and one third of its annotated genes are still hypothetical. To identify genetic determinants expressed under a variety of environmental factors, we applied strand-specific RNA-sequencing, comparing the SOLiD and Illumina systems. Results: Transcriptomes of EHEC were sequenced under 11 different biotic and abiotic conditions: LB medium at pH4, pH7, pH9, or at 15°C; LB with nitrite or trimethoprim-sulfamethoxazole; LB-agar surface, M9 minimal medium, spinach leaf juice, surface of living radish sprouts, and cattle feces. Of 5379 annotated genes in strain EDL933 (genome and plasmid), a surprising minority of only 144 had null sequencing reads under all conditions. We therefore developed a statistical method to distinguish weakly transcribed genes from background transcription. We find that 96% of all genes and 91.5% of the hypothetical genes exhibit a significant transcriptional signal under at least one condition. Comparing SOLiD and Illumina systems, we find a high correlation between both approaches for fold-changes of the induced or repressed genes. The pathogenicity island LEE showed highest transcriptional activity in LB medium, minimal medium, and after treatment with antibiotics. -

Determining the Effect of Small Doses of Fructose and Its Epimers on Glycemic Control
Determining the Effect of Small Doses of Fructose and its Epimers on Glycemic Control by Jarvis Clyde Noronha A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Nutritional Sciences University of Toronto © Copyright by Jarvis Clyde Noronha 2017 ii Determining the Effect of Small Doses of Fructose and its Epimers on Glycemic Control Jarvis Clyde Noronha Master of Science Department of Nutritional Sciences University of Toronto 2017 Abstract Given that sugars have emerged as the dominant nutrient of concern in diabetes, there is a need for the development of alternative sweeteners. To assess the role of small doses (5g, 10g) of fructose and allulose on postprandial glucose metabolism, we conducted an acute randomized controlled trial in individuals with type 2 diabetes. We found that small doses of allulose, but not fructose, modestly reduced the postprandial glycemic response to a 75g oral glucose load. To assess whether low-dose (< 50-g/day) fructose and all its epimers (allulose, tagatose and sorbose) lead to sustainable improvements in long-term glycemic control, we conducted a systematic review and meta-analysis of controlled feeding trials. The available evidence suggested that fructose and tagatose led to significant reductions in HbA1c and fasting glucose. Our findings highlight the need for long-term randomized controlled trials to confirm the viability of fructose and its epimers as alternative sweeteners. iii Table of Contents Contents Abstract ........................................................................................................................... -

Non-Homologous Isofunctional Enzymes: a Systematic Analysis Of
Omelchenko et al. Biology Direct 2010, 5:31 http://www.biology-direct.com/content/5/1/31 RESEARCH Open Access Non-homologousResearch isofunctional enzymes: A systematic analysis of alternative solutions in enzyme evolution Marina V Omelchenko, Michael Y Galperin*, Yuri I Wolf and Eugene V Koonin Abstract Background: Evolutionarily unrelated proteins that catalyze the same biochemical reactions are often referred to as analogous - as opposed to homologous - enzymes. The existence of numerous alternative, non-homologous enzyme isoforms presents an interesting evolutionary problem; it also complicates genome-based reconstruction of the metabolic pathways in a variety of organisms. In 1998, a systematic search for analogous enzymes resulted in the identification of 105 Enzyme Commission (EC) numbers that included two or more proteins without detectable sequence similarity to each other, including 34 EC nodes where proteins were known (or predicted) to have distinct structural folds, indicating independent evolutionary origins. In the past 12 years, many putative non-homologous isofunctional enzymes were identified in newly sequenced genomes. In addition, efforts in structural genomics resulted in a vastly improved structural coverage of proteomes, providing for definitive assessment of (non)homologous relationships between proteins. Results: We report the results of a comprehensive search for non-homologous isofunctional enzymes (NISE) that yielded 185 EC nodes with two or more experimentally characterized - or predicted - structurally unrelated proteins. Of these NISE sets, only 74 were from the original 1998 list. Structural assignments of the NISE show over-representation of proteins with the TIM barrel fold and the nucleotide-binding Rossmann fold. From the functional perspective, the set of NISE is enriched in hydrolases, particularly carbohydrate hydrolases, and in enzymes involved in defense against oxidative stress. -

Preclinical Evaluation of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma by Andrea Shergalis
Preclinical Evaluation of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma By Andrea Shergalis A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Medicinal Chemistry) in the University of Michigan 2020 Doctoral Committee: Professor Nouri Neamati, Chair Professor George A. Garcia Professor Peter J. H. Scott Professor Shaomeng Wang Andrea G. Shergalis [email protected] ORCID 0000-0002-1155-1583 © Andrea Shergalis 2020 All Rights Reserved ACKNOWLEDGEMENTS So many people have been involved in bringing this project to life and making this dissertation possible. First, I want to thank my advisor, Prof. Nouri Neamati, for his guidance, encouragement, and patience. Prof. Neamati instilled an enthusiasm in me for science and drug discovery, while allowing me the space to independently explore complex biochemical problems, and I am grateful for his kind and patient mentorship. I also thank my committee members, Profs. George Garcia, Peter Scott, and Shaomeng Wang, for their patience, guidance, and support throughout my graduate career. I am thankful to them for taking time to meet with me and have thoughtful conversations about medicinal chemistry and science in general. From the Neamati lab, I would like to thank so many. First and foremost, I have to thank Shuzo Tamara for being an incredible, kind, and patient teacher and mentor. Shuzo is one of the hardest workers I know. In addition to a strong work ethic, he taught me pretty much everything I know and laid the foundation for the article published as Chapter 3 of this dissertation. The work published in this dissertation really began with the initial identification of PDI as a target by Shili Xu, and I am grateful for his advice and guidance (from afar!). -

FDA Finalizes Allulose Guidance and Requests Information on Other Sugars Metabolized Differently Than Traditional Sugars
FDA Finalizes Allulose Guidance and Requests Information on Other Sugars Metabolized Differently Than Traditional Sugars October 19, 2020 Food, Drug, and Device FDA has taken two notable actions regarding the sugars declaration in the Nutrition Facts Label (NFL) and Supplement Facts Label (SFL). On Friday, the agency released a final guidance regarding the declaration of allulose, confirming that this monosaccharide need not be included in the declaration of “Total Sugars” or “Added Sugars,” though it must be included in the “Total Carbohydrates” declaration in the NFL. Today, FDA published a Federal Register notice requesting information about and comments on the nutrition labeling of other sugars that are metabolized differently than traditional sugars. We briefly summarize both documents below to help inform stakeholder comments on the notice, which are due to FDA by December 18, 2020. Allulose Final Guidance Allulose, or D-psicose, is a monosaccharide that can be used as a substitute for traditional sugar in food and beverage products. For purposes of nutrition labeling, FDA has generally 1 defined nutrients based on their chemical structure.0F Accordingly, when FDA updated its NFL and SFL regulations in 2016, the agency reiterated the definition of “Total Sugars” as the sum of 2 all free monosaccharides and disaccharides (e.g. glucose, fructose, and sucrose).1F FDA also added to these regulations a definition of “Added Sugars” – sugars added during the processing 3 of food, or packaged as such – and required their declaration in the NFL and SFL.2F Although the agency recognized that there are sugars that are metabolized differently than traditional sugars, FDA did not make a determination at that time as to whether allulose should be excluded from “Total Carbohydrate,” “Total Sugars,” or “Added Sugars” Declarations. -

Effects of D-Allulose on Glucose Tolerance and Insulin Response to A
Clinical care/Education/Nutrition Open access Original research BMJ Open Diab Res Care: first published as 10.1136/bmjdrc-2020-001939 on 26 February 2021. Downloaded from Effects of D- allulose on glucose tolerance and insulin response to a standard oral sucrose load: results of a prospective, randomized, crossover study Francesco Franchi ,1 Dmitry M Yaranov,1 Fabiana Rollini,1 Andrea Rivas,1 Jose Rivas Rios,1 Latonya Been,1 Yuma Tani,2 Masaaki Tokuda,3 Tetsuo Iida,2 Noriko Hayashi,2 Dominick J Angiolillo,1 Arshag D Mooradian1 To cite: Franchi F, Yaranov DM, ABSTRACT Rollini F, et al. Effects of D- Introduction Current dietary guidelines recommend Significance of this study allulose on glucose tolerance limiting sugar intake for the prevention of diabetes and insulin response to a mellitus (DM). Reduction in sugar intake may require sugar What is already known about this subject? standard oral sucrose load: substitutes. Among these, D- allulose is a non- calorie rare ► D- allulose is defined one of the rare sugars, results of a prospective, which has been shown in animal and clinical randomized, crossover study. monosaccharide with 70% sweetness of sucrose, which has shown anti- DM effects in Asian populations. However, studies, conducted mostly in Asian populations, BMJ Open Diab Res Care to have postprandial plasma glucose suppres- 2021;9:e001939. doi:10.1136/ there is limited data on the effects of D- allulose in other sive effects with antiobesity and antidiabetic bmjdrc-2020-001939 populations, including Westerners. Research design and methods This was a prospective, effects. copyright. randomized, double- blind, placebo- controlled, crossover What are the new findings? ► Supplemental material is study conducted in 30 subjects without DM. -

Recent Advances in Trypanosomatid Research: Genome Organization, Expression, Metabolism, Taxonomy and Evolution
Parasitology Recent advances in trypanosomatid research: genome organization, expression, metabolism, cambridge.org/par taxonomy and evolution 1 2 3,4 5,6 Review Dmitri A. Maslov , Fred R. Opperdoes , Alexei Y. Kostygov , Hassan Hashimi , Julius Lukeš5,6 and Vyacheslav Yurchenko3,5,7 Cite this article: Maslov DA, Opperdoes FR, Kostygov AY, Hashimi H, Lukeš J, Yurchenko V 1Department of Molecular, Cell and Systems Biology, University of California – Riverside, Riverside, California, USA; (2018). Recent advances in trypanosomatid 2de Duve Institute, Université Catholique de Louvain, Brussels, Belgium; 3Life Science Research Centre, Faculty of research: genome organization, expression, 4 metabolism, taxonomy and evolution. Science, University of Ostrava, Ostrava, Czech Republic; Zoological Institute of the Russian Academy of Sciences, 5 Parasitology 1–27. https://doi.org/10.1017/ St. Petersburg, Russia; Biology Centre, Institute of Parasitology, Czech Academy of Sciences, České Budejovice 6 S0031182018000951 (Budweis), Czech Republic; University of South Bohemia, Faculty of Sciences, České Budejovice (Budweis), Czech Republic and 7Martsinovsky Institute of Medical Parasitology, Tropical and Vector Borne Diseases, Sechenov Received: 30 January 2018 University, Moscow, Russia Revised: 23 April 2018 Accepted: 23 April 2018 Abstract Key words: Unicellular flagellates of the family Trypanosomatidae are obligatory parasites of inverte- Gene exchange; kinetoplast; metabolism; molecular and cell biology; taxonomy; brates, vertebrates and plants. Dixenous species are aetiological agents of a number of diseases trypanosomatidae in humans, domestic animals and plants. Their monoxenous relatives are restricted to insects. Because of the high biological diversity, adaptability to dramatically different environmental Author for correspondence: conditions, and omnipresence, these protists have major impact on all biotic communities Vyacheslav Yurchenko, E-mail: vyacheslav. -

Enzymatic Synthesis of L-Fucose from L-Fuculose Using a Fucose Isomerase
Kim et al. Biotechnol Biofuels (2019) 12:282 https://doi.org/10.1186/s13068-019-1619-0 Biotechnology for Biofuels RESEARCH Open Access Enzymatic synthesis of L-fucose from L-fuculose using a fucose isomerase from Raoultella sp. and the biochemical and structural analyses of the enzyme In Jung Kim1†, Do Hyoung Kim1†, Ki Hyun Nam1,2 and Kyoung Heon Kim1* Abstract Background: L-Fucose is a rare sugar with potential uses in the pharmaceutical, cosmetic, and food industries. The enzymatic approach using L-fucose isomerase, which interconverts L-fucose and L-fuculose, can be an efcient way of producing L-fucose for industrial applications. Here, we performed biochemical and structural analyses of L-fucose isomerase identifed from a novel species of Raoultella (RdFucI). Results: RdFucI exhibited higher enzymatic activity for L-fuculose than for L-fucose, and the rate for the reverse reaction of converting L-fuculose to L-fucose was higher than that for the forward reaction of converting L-fucose to L-fuculose. In the equilibrium mixture, a much higher proportion of L-fucose (~ ninefold) was achieved at 30 °C and pH 7, indicating that the enzyme-catalyzed reaction favors the formation of L-fucose from L-fuculose. When biochemical analysis was conducted using L-fuculose as the substrate, the optimal conditions for RdFucI activity were determined to be 40 °C and pH 10. However, the equilibrium composition was not afected by reaction temperature in the range 2 of 30 to 50 °C. Furthermore, RdFucI was found to be a metalloenzyme requiring Mn + as a cofactor. The comparative crystal structural analysis of RdFucI revealed the distinct conformation of α7–α8 loop of RdFucI. -
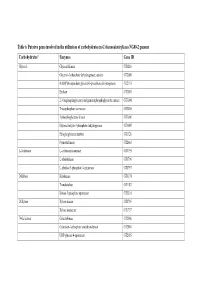
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

SUPPY Liglucosexlmtdh
US 20100314248A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0314248 A1 Worden et al. (43) Pub. Date: Dec. 16, 2010 (54) RENEWABLE BOELECTRONIC INTERFACE Publication Classification FOR ELECTROBOCATALYTC REACTOR (51) Int. Cl. (76) Inventors: Robert M. Worden, Holt, MI (US); C25B II/06 (2006.01) Brian L. Hassler, Lake Orion, MI C25B II/2 (2006.01) (US); Lawrence T. Drzal, Okemos, GOIN 27/327 (2006.01) MI (US); Ilsoon Lee, Okemo s, MI BSD L/04 (2006.01) (US) C25B 9/00 (2006.01) (52) U.S. Cl. ............... 204/403.14; 204/290.11; 204/400; Correspondence Address: 204/290.07; 427/458; 204/252: 977/734; PRICE HENEVELD COOPER DEWITT & LIT 977/742 TON, LLP 695 KENMOOR, S.E., PO BOX 2567 (57) ABSTRACT GRAND RAPIDS, MI 495.01 (US) An inexpensive, easily renewable bioelectronic device useful for bioreactors, biosensors, and biofuel cells includes an elec (21) Appl. No.: 12/766,169 trically conductive carbon electrode and a bioelectronic inter face bonded to a surface of the electrically conductive carbon (22) Filed: Apr. 23, 2010 electrode, wherein the bioelectronic interface includes cata lytically active material that is electrostatically bound directly Related U.S. Application Data or indirectly to the electrically conductive carbon electrode to (60) Provisional application No. 61/172,337, filed on Apr. facilitate easy removal upon a change in pH, thereby allowing 24, 2009. easy regeneration of the bioelectronic interface. 7\ POWER 1 - SUPPY|- LIGLUCOSEXLMtDH?till pi 6.0 - esses&aaaas-exx-xx-xx-xx-xxxxixax-e- Patent Application Publication Dec. 16, 2010 Sheet 1 of 18 US 2010/0314248 A1 Potential (nV) Patent Application Publication Dec. -

The Food Lawyers® Respectfully Request That FDA Implements the Following
December 7, 2020 Dockets Management Staff (HFA-305) Filed Electronically Food and Drug Administration https://www.regulations.gov Re: Sugars Metabolized Differently than Traditional Sugars (FDA-2020-N-1359) Ladies and Gentlemen: One Page Executive Summary FDA’s seeks information to “… promote the public health and help consumers make informed dietary decisions” regarding sugars that are metabolized differently than traditional sugars. Given the nation’s battles with diabetes and obesity, and the benefits that non-traditional sugars can offer in these battles, the Agency’s stated public policy goal goes to the very heart of American consumers’ health. This laudatory public policy’s realization is complicated by a lack of consumer awareness of how some sugars are metabolized differently than others. In an effort to answer the questions posed by the Agency regarding the treatment of Sugars that Are Metabolized Differently Than Traditional Sugars, we suggest that the Agency adapt a mechanism that will seek to harmonize the public policy of promoting public health with consumers’ lack of awareness of sugars that are metabolized differently than sucrose. In particular, we suggest that FDA should consider the following: 1. Establish a new category of sugars called Rare Sugars that exhibit the following characteristics: a. Are naturally occurring b. Impart a sweet taste that is at least 50% the sweetness of sucrose c. 2.0 kcal/g or less. d. Resulting pH of 6.0 or greater of dental plaque after consumption. e. No or low glycemic response. f. No or low insulinemic response. 2. Exclude Rare Sugars from “Total Sugars” and “Added Sugars” declarations to stimulate their deployment by industry and consumption by the public.