Predictions of Response to Cancer Immunotherapy Via Tumour Mutational Burden and Genomic Resistance Markers Jacob Bradley1 and Nirmesh Patel Phd2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Incorporating Genetic Networks Into Case-Control Association Studies with High-Dimensional DNA Methylation Data Kipoong Kim and Hokeun Sun*
Kim and Sun BMC Bioinformatics (2019) 20:510 https://doi.org/10.1186/s12859-019-3040-x METHODOLOGY ARTICLE Open Access Incorporating genetic networks into case-control association studies with high-dimensional DNA methylation data Kipoong Kim and Hokeun Sun* Abstract Background: In human genetic association studies with high-dimensional gene expression data, it has been well known that statistical selection methods utilizing prior biological network knowledge such as genetic pathways and signaling pathways can outperform other methods that ignore genetic network structures in terms of true positive selection. In recent epigenetic research on case-control association studies, relatively many statistical methods have been proposed to identify cancer-related CpG sites and their corresponding genes from high-dimensional DNA methylation array data. However, most of existing methods are not designed to utilize genetic network information although methylation levels between linked genes in the genetic networks tend to be highly correlated with each other. Results: We propose new approach that combines data dimension reduction techniques with network-based regularization to identify outcome-related genes for analysis of high-dimensional DNA methylation data. In simulation studies, we demonstrated that the proposed approach overwhelms other statistical methods that do not utilize genetic network information in terms of true positive selection. We also applied it to the 450K DNA methylation array data of the four breast invasive carcinoma cancer subtypes from The Cancer Genome Atlas (TCGA) project. Conclusions: The proposed variable selection approach can utilize prior biological network information for analysis of high-dimensional DNA methylation array data. It first captures gene level signals from multiple CpG sites using data a dimension reduction technique and then performs network-based regularization based on biological network graph information. -

PDF Datasheet
Product Datasheet Recombinant Human OR5P2 GST (N-Term) Protein H00120065-P01-2ug Unit Size: 2 ug Store at -80C. Avoid freeze-thaw cycles. Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/H00120065-P01 Updated 11/4/2020 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/H00120065-P01 Page 1 of 3 v.20.1 Updated 11/4/2020 H00120065-P01-2ug Recombinant Human OR5P2 GST (N-Term) Protein Product Information Unit Size 2 ug Concentration Please see the vial label for concentration. If unlisted please contact technical services. Storage Store at -80C. Avoid freeze-thaw cycles. Preservative No Preservative Purity >80% by SDS-PAGE and Coomassie blue staining Buffer 50 mM Tris-HCl, 10 mM reduced Glutathione, pH 8.0 in the elution buffer. Target Molecular Weight 62.2 kDa Product Description Description A recombinant protein with GST-tag at N-terminal corresponding to the amino acids 1 - 322 of Human OR5P2 full-length ORF Source: Wheat Germ (in vitro) Amino Acid Sequence: MNSLKDGNHTALTGFILLGLTDDPILRVILFMIILSGNLSIIILIRISSQLHHPMYFFL SHLAFADMAYSSSVTPNMLVNFLVERNTVSYLGCAIQLGSAAFFATVECVLLAA MAYDRFVAICSPLLYSTKMSTQVSVQLLLVVYIAGFLIAVSYTTSFYFLLFCGPN QVNHFFCDFAPLLELSCSDISVSTVVLSFSSGSIIVVTVCVIAVCYIYILITILKMRS TEGHHKAFSTCTSHLTVVTLFYGTITFIYVMPNFSYSTDQNKVVSVLYTVVIPML NPLIYSLRNKEIKGALKRELVRKILSHDACYFSRTSNNDIT Gene ID 120065 Gene Symbol OR5P2 Species Human Preparation Method in vitro wheat germ expression system Details of Functionality This protein was produced in an in vitro wheat germ expression system that should preserve correct conformational folding that is necessary for biological function. While it is possible that this protein could display some level of activity, the functionality of this protein has not been explicitly measured or validated. -

Multiple Loci Modulate Opioid Therapy Response for Cancer Pain
Published OnlineFirst May 27, 2011; DOI: 10.1158/1078-0432.CCR-10-3028 Clinical Cancer Predictive Biomarkers and Personalized Medicine Research Multiple Loci Modulate Opioid Therapy Response for Cancer Pain Antonella Galvan1, Frank Skorpen2,Pal Klepstad2,3, Anne Kari Knudsen2, Torill Fladvad2, Felicia S. Falvella1, Alessandra Pigni1, Cinzia Brunelli1, Augusto Caraceni1,2, Stein Kaasa2,4, and Tommaso A. Dragani1 Abstract Purpose: Patients treated with opioid drugs for cancer pain experience different relief responses, raising the possibility that genetic factors play a role in opioid therapy outcome. In this study, we tested the hypothesis that genetic variations may control individual response to opioid drugs in cancer patients. Experimental Design: We tested 1 million single-nucleotide polymorphisms (SNP) in European cancer patients, selected in a first series, for extremely poor (pain relief 40%; n ¼ 145) or good (pain relief 90%; n ¼ 293) responses to opioid therapy using a DNA-pooling approach. Candidate SNPs identified by SNP- array were genotyped in individual samples constituting DNA pools as well as in a second series of 570 patients. Results: Association analysis in 1,008 cancer patients identified eight SNPs significantly associated with À pain relief at a statistical threshold of P < 1.0 Â 10 3, with rs12948783, upstream of the RHBDF2 gene, À showing the best statistical association (P ¼ 8.1 Â 10 9). Functional annotation analysis of SNP-tagged genes suggested the involvement of genes acting on processes of the neurologic system. Conclusion: Our results indicate that the identified SNP panel can modulate the response of cancer patients to opioid therapy and may provide a new tool for personalized therapy of cancer pain. -

Sex-Specific Transcriptome Differences in Human Adipose
G C A T T A C G G C A T genes Article Sex-Specific Transcriptome Differences in Human Adipose Mesenchymal Stem Cells 1, 2, 3 1,3 Eva Bianconi y, Raffaella Casadei y , Flavia Frabetti , Carlo Ventura , Federica Facchin 1,3,* and Silvia Canaider 1,3 1 National Laboratory of Molecular Biology and Stem Cell Bioengineering of the National Institute of Biostructures and Biosystems (NIBB)—Eldor Lab, at the Innovation Accelerator, CNR, Via Piero Gobetti 101, 40129 Bologna, Italy; [email protected] (E.B.); [email protected] (C.V.); [email protected] (S.C.) 2 Department for Life Quality Studies (QuVi), University of Bologna, Corso D’Augusto 237, 47921 Rimini, Italy; [email protected] 3 Department of Experimental, Diagnostic and Specialty Medicine (DIMES), University of Bologna, Via Massarenti 9, 40138 Bologna, Italy; fl[email protected] * Correspondence: [email protected]; Tel.: +39-051-2094114 These authors contributed equally to this work. y Received: 1 July 2020; Accepted: 6 August 2020; Published: 8 August 2020 Abstract: In humans, sexual dimorphism can manifest in many ways and it is widely studied in several knowledge fields. It is increasing the evidence that also cells differ according to sex, a correlation still little studied and poorly considered when cells are used in scientific research. Specifically, our interest is on the sex-related dimorphism on the human mesenchymal stem cells (hMSCs) transcriptome. A systematic meta-analysis of hMSC microarrays was performed by using the Transcriptome Mapper (TRAM) software. This bioinformatic tool was used to integrate and normalize datasets from multiple sources and allowed us to highlight chromosomal segments and genes differently expressed in hMSCs derived from adipose tissue (hADSCs) of male and female donors. -

The Mutational Landscape of Human Olfactory G Protein-Coupled Receptors
Jimenez et al. BMC Biology (2021) 19:21 https://doi.org/10.1186/s12915-021-00962-0 RESEARCH ARTICLE Open Access The mutational landscape of human olfactory G protein-coupled receptors Ramón Cierco Jimenez1,2, Nil Casajuana-Martin1, Adrián García-Recio1, Lidia Alcántara1, Leonardo Pardo1, Mercedes Campillo1 and Angel Gonzalez1* Abstract Background: Olfactory receptors (ORs) constitute a large family of sensory proteins that enable us to recognize a wide range of chemical volatiles in the environment. By contrast to the extensive information about human olfactory thresholds for thousands of odorants, studies of the genetic influence on olfaction are limited to a few examples. To annotate on a broad scale the impact of mutations at the structural level, here we analyzed a compendium of 119,069 natural variants in human ORs collected from the public domain. Results: OR mutations were categorized depending on their genomic and protein contexts, as well as their frequency of occurrence in several human populations. Functional interpretation of the natural changes was estimated from the increasing knowledge of the structure and function of the G protein-coupled receptor (GPCR) family, to which ORs belong. Our analysis reveals an extraordinary diversity of natural variations in the olfactory gene repertoire between individuals and populations, with a significant number of changes occurring at the structurally conserved regions. A particular attention is paid to mutations in positions linked to the conserved GPCR activation mechanism that could imply phenotypic variation in the olfactory perception. An interactive web application (hORMdb, Human Olfactory Receptor Mutation Database) was developed for the management and visualization of this mutational dataset. -

Olfactory Receptors in Non-Chemosensory Tissues
BMB Reports Invited Mini Review Olfactory receptors in non-chemosensory tissues NaNa Kang & JaeHyung Koo* Department of Brain Science, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 711-873, Korea Olfactory receptors (ORs) detect volatile chemicals that lead to freezing behavior (3-5). the initial perception of smell in the brain. The olfactory re- ORs are localized in the cilia of olfactory sensory neurons ceptor (OR) is the first protein that recognizes odorants in the (OSNs) in the olfactory epithelium (OE) and are activated by olfactory signal pathway and it is present in over 1,000 genes chemical cues, typically odorants at the molecular level, in mice. It is also the largest member of the G protein-coupled which lead to the perception of smell in the brain (6). receptors (GPCRs). Most ORs are extensively expressed in the Tremendous research was conducted since Buck and Axel iso- nasal olfactory epithelium where they perform the appropriate lated ORs as an OE-specific expression in 1991 (7). OR genes, physiological functions that fit their location. However, recent the largest family among the G protein-coupled receptors whole-genome sequencing shows that ORs have been found (GPCRs) (8), constitute more than 1,000 genes on the mouse outside of the olfactory system, suggesting that ORs may play chromosome (9, 10) and more than 450 genes in the human an important role in the ectopic expression of non-chemo- genome (11, 12). sensory tissues. The ectopic expressions of ORs and their phys- Odorant activation shows a distinct signal transduction iological functions have attracted more attention recently since pathway for odorant perception. -
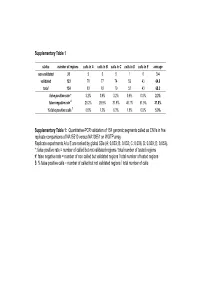
Quantitative-PCR Validation of 154 Genomic Segments Called As Cnvs in Five Replicat
Supplementary Table 1 status number of regions calls in A calls in B calls in C calls in D calls in E average non validated 31 5 6 5 1 0 3.4 validated 123 78 77 74 52 43 64.8 total 154 83 83 79 53 43 68.2 false positive rate * 3.2% 3.9% 3.2% 0.6% 0.0% 2.2% false negative rate # 29.2% 29.9% 31.8% 46.1% 51.9% 37.8% % false positive calls $ 6.0% 7.2% 6.3% 1.9% 0.0% 5.0% Supplementary Table 1: Quantitative-PCR validation of 154 genomic segments called as CNVs in five replicate comparisons of NA15510 versus NA10851 on WGTP array Replicate experiments A to E are ranked by global SDe (A: 0.033; B: 0.033; C: 0.036; D: 0.039; E: 0.053). *: false positive rate = number of called but not validated regions / total number of tested regions #: false negative rate = number of non called but validated regions / total number of tested regions $: % false positive calls = number of called but not validated regions / total number of calls False positive estimates for 500K EA CNV calls Total Rep1 Rep2 Rep3 Avg (unique) Validated 33 28 32 31 38 Not validated 2 2 2 2 5 Total 35 30 34 33 43 % False positive 5.71% 6.67% 5.88% 6.09% - % False negative 13.16% 26.32% 15.79% 18.42% - Supplementary Table 2A : Quantitative PCR validation of 43 unique CNV regions called as CNVs in three replicate comparisons of NA15510 versus NA10851 using the 500K EA array. -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

IPP17 Carcinoma IPMN Gene Symbol Transcript Accession Nucleotide
IPP17 Carcinoma IPMN Gene Transcript Amino Gene Transcript Amino Allele Nucleotide change Consequence Allele Frequency Nucleotide change Consequence Symbol Accession Acid Symbol Accession Acid Frequency ACAP2 CCDS33924.1 chr3: 195063234 C>T C165Y Missense 19 ADCY8 CCDS33924.1 chr8: 131964235 C>T V374M Missense 29 ADAMTS1 CCDS33524.1 chr21: 28212054 T>G E627A Missense 11 ASH1L CCDS6363.1 chr1: 155307924 T>C N2920S Missense 16 ADCY8 CCDS6363.1 chr8: 131964235 C>T V374M Missense 36 CLNK CCDS1113.2 chr4: 10542196 G>T P175H Missense 14 AR CCDS14387.1 chrX: 66765158 T>A L57Q Missense 15 CLU CCDS47024.1 chr1: 155307924 T>C R127H Missense 23 ARL11 CCDS9419.1 chr13: 50204931 delCTT F117del In-frame deletion 10 CPN1 CCDS47832.1 chr10: 101816891 T>A Y297F In-frame deletion 13 ASH1L CCDS1113.2 chr1: 155307924 T>C N2920S Missense 16 CSRNP2 CCDS7486.1 chr12: 51458139 delTCT E340del In-frame deletion 13 C7orf26 CCDS5353.1 chr7: 6647641 C>T A400V Missense 13 CTNNB1 CCDS8807.1 chr3: 41266136 T>C S45P Missense 11 CLNK CCDS47024.1 chr4: 10542196 G>T P175H Missense 14 ERF CCDS2694.1 chr19: 42752892 G>A R458C Missense 15 COMT CCDS13770.1 chr22: 19951169 G>A V124M Missense 11 FBP2 CCDS12600.1 chr9: 97325736 C>A S238I Missense 13 CORIN CCDS3477.1 chr4: 47645243 G>C A663G Missense 14 HOXB3 CCDS6711.1 chr17: 46629769 C>T G23D Missense 13 CPN1 CCDS8807.1 chr10: 101816891 T>A Y297F In-frame deletion 9 KRTAP13 CCDS11528.1 chr21: 31797951 T>C T94A Missense 14 CSRNP2 CCDS2694.1 chr12: 51458139 delTCT E340del In-frame deletion 17 MAGEB4 CCDS13591.1 chrX: 30260590 C>T -

Positive Selection, Relaxation, and Acceleration in the Evolution of the Human and Chimp Genome
Positive Selection, Relaxation, and Acceleration in the Evolution of the Human and Chimp Genome Leonardo Arbiza1, Joaquı´n Dopazo2, Herna´n Dopazo1* 1 Pharmacogenomics and Comparative Genomics Unit, Centro de Investigacio´nPrı´ncipe Felipe (CIPF), Valencia, Spain, 2 Functional Genomics Unit, Bioinformatics Department, Centro de Investigacio´nPrı´ncipe Felipe (CIPF), Valencia, Spain For years evolutionary biologists have been interested in searching for the genetic bases underlying humanness. Recent efforts at a large or a complete genomic scale have been conducted to search for positively selected genes in human and in chimp. However, recently developed methods allowing for a more sensitive and controlled approach in the detection of positive selection can be employed. Here, using 13,198 genes, we have deduced the sets of genes involved in rate acceleration, positive selection, and relaxation of selective constraints in human, in chimp, and in their ancestral lineage since the divergence from murids. Significant deviations from the strict molecular clock were observed in 469 human and in 651 chimp genes. The more stringent branch-site test of positive selection detected 108 human and 577 chimp positively selected genes. An important proportion of the positively selected genes did not show a significant acceleration in rates, and similarly, many of the accelerated genes did not show significant signals of positive selection. Functional differentiation of genes under rate acceleration, positive selection, and relaxation was not statistically significant between human and chimp with the exception of terms related to G-protein coupled receptors and sensory perception. Both of these were over-represented under relaxation in human in relation to chimp. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.