Multiple Loci Modulate Opioid Therapy Response for Cancer Pain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sex-Specific Transcriptome Differences in Human Adipose
G C A T T A C G G C A T genes Article Sex-Specific Transcriptome Differences in Human Adipose Mesenchymal Stem Cells 1, 2, 3 1,3 Eva Bianconi y, Raffaella Casadei y , Flavia Frabetti , Carlo Ventura , Federica Facchin 1,3,* and Silvia Canaider 1,3 1 National Laboratory of Molecular Biology and Stem Cell Bioengineering of the National Institute of Biostructures and Biosystems (NIBB)—Eldor Lab, at the Innovation Accelerator, CNR, Via Piero Gobetti 101, 40129 Bologna, Italy; [email protected] (E.B.); [email protected] (C.V.); [email protected] (S.C.) 2 Department for Life Quality Studies (QuVi), University of Bologna, Corso D’Augusto 237, 47921 Rimini, Italy; [email protected] 3 Department of Experimental, Diagnostic and Specialty Medicine (DIMES), University of Bologna, Via Massarenti 9, 40138 Bologna, Italy; fl[email protected] * Correspondence: [email protected]; Tel.: +39-051-2094114 These authors contributed equally to this work. y Received: 1 July 2020; Accepted: 6 August 2020; Published: 8 August 2020 Abstract: In humans, sexual dimorphism can manifest in many ways and it is widely studied in several knowledge fields. It is increasing the evidence that also cells differ according to sex, a correlation still little studied and poorly considered when cells are used in scientific research. Specifically, our interest is on the sex-related dimorphism on the human mesenchymal stem cells (hMSCs) transcriptome. A systematic meta-analysis of hMSC microarrays was performed by using the Transcriptome Mapper (TRAM) software. This bioinformatic tool was used to integrate and normalize datasets from multiple sources and allowed us to highlight chromosomal segments and genes differently expressed in hMSCs derived from adipose tissue (hADSCs) of male and female donors. -

The Mutational Landscape of Human Olfactory G Protein-Coupled Receptors
Jimenez et al. BMC Biology (2021) 19:21 https://doi.org/10.1186/s12915-021-00962-0 RESEARCH ARTICLE Open Access The mutational landscape of human olfactory G protein-coupled receptors Ramón Cierco Jimenez1,2, Nil Casajuana-Martin1, Adrián García-Recio1, Lidia Alcántara1, Leonardo Pardo1, Mercedes Campillo1 and Angel Gonzalez1* Abstract Background: Olfactory receptors (ORs) constitute a large family of sensory proteins that enable us to recognize a wide range of chemical volatiles in the environment. By contrast to the extensive information about human olfactory thresholds for thousands of odorants, studies of the genetic influence on olfaction are limited to a few examples. To annotate on a broad scale the impact of mutations at the structural level, here we analyzed a compendium of 119,069 natural variants in human ORs collected from the public domain. Results: OR mutations were categorized depending on their genomic and protein contexts, as well as their frequency of occurrence in several human populations. Functional interpretation of the natural changes was estimated from the increasing knowledge of the structure and function of the G protein-coupled receptor (GPCR) family, to which ORs belong. Our analysis reveals an extraordinary diversity of natural variations in the olfactory gene repertoire between individuals and populations, with a significant number of changes occurring at the structurally conserved regions. A particular attention is paid to mutations in positions linked to the conserved GPCR activation mechanism that could imply phenotypic variation in the olfactory perception. An interactive web application (hORMdb, Human Olfactory Receptor Mutation Database) was developed for the management and visualization of this mutational dataset. -

IPP17 Carcinoma IPMN Gene Symbol Transcript Accession Nucleotide
IPP17 Carcinoma IPMN Gene Transcript Amino Gene Transcript Amino Allele Nucleotide change Consequence Allele Frequency Nucleotide change Consequence Symbol Accession Acid Symbol Accession Acid Frequency ACAP2 CCDS33924.1 chr3: 195063234 C>T C165Y Missense 19 ADCY8 CCDS33924.1 chr8: 131964235 C>T V374M Missense 29 ADAMTS1 CCDS33524.1 chr21: 28212054 T>G E627A Missense 11 ASH1L CCDS6363.1 chr1: 155307924 T>C N2920S Missense 16 ADCY8 CCDS6363.1 chr8: 131964235 C>T V374M Missense 36 CLNK CCDS1113.2 chr4: 10542196 G>T P175H Missense 14 AR CCDS14387.1 chrX: 66765158 T>A L57Q Missense 15 CLU CCDS47024.1 chr1: 155307924 T>C R127H Missense 23 ARL11 CCDS9419.1 chr13: 50204931 delCTT F117del In-frame deletion 10 CPN1 CCDS47832.1 chr10: 101816891 T>A Y297F In-frame deletion 13 ASH1L CCDS1113.2 chr1: 155307924 T>C N2920S Missense 16 CSRNP2 CCDS7486.1 chr12: 51458139 delTCT E340del In-frame deletion 13 C7orf26 CCDS5353.1 chr7: 6647641 C>T A400V Missense 13 CTNNB1 CCDS8807.1 chr3: 41266136 T>C S45P Missense 11 CLNK CCDS47024.1 chr4: 10542196 G>T P175H Missense 14 ERF CCDS2694.1 chr19: 42752892 G>A R458C Missense 15 COMT CCDS13770.1 chr22: 19951169 G>A V124M Missense 11 FBP2 CCDS12600.1 chr9: 97325736 C>A S238I Missense 13 CORIN CCDS3477.1 chr4: 47645243 G>C A663G Missense 14 HOXB3 CCDS6711.1 chr17: 46629769 C>T G23D Missense 13 CPN1 CCDS8807.1 chr10: 101816891 T>A Y297F In-frame deletion 9 KRTAP13 CCDS11528.1 chr21: 31797951 T>C T94A Missense 14 CSRNP2 CCDS2694.1 chr12: 51458139 delTCT E340del In-frame deletion 17 MAGEB4 CCDS13591.1 chrX: 30260590 C>T -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -
UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Systems Biology of Liver Regeneration and Pathologies Permalink https://escholarship.org/uc/item/05d214b4 Author Min, Jun SungJun Publication Date 2015 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Systems Biology of Liver Regeneration and Pathologies A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioengineering by Jun SungJun Min Committee in charge: Professor Shankar Subramaniam, Chair Professor Pedro Cabrales Professor Daniel Tartakovsky Professor Shyni Varghese Professor Yingxiao Wang 2015 Copyright Jun SungJun Min, 2015 All rights reserved. The Dissertation of Jun SungJun Min is approved, and it is acceptable in quality and form for publication on microfilm and electronically: ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ Chair University of California, San Diego 2015 iii DEDICATION To my friends and family With their love and support iv TABLE OF CONTENTS Signature Page ..................................................................................................... iii Dedication ........................................................................................................... -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

Supplementary Tables
Supplementary Tables Supplementary Table 1. Differentially methylated genes in correlation with their expression pattern in the A4 progression model A. Hypomethylated–upregulated Genes (n= 76) ALOX5 RRAD RTN4R DSCR6 FGFR3 HTR7 WNT3A POGK PLCD3 ALPPL2 RTEL1 SEMA3B DUSP5 FOSB ITGB4 MEST PPL PSMB8 ARHGEF4 BST2 SEMA7A SLC12A7 FOXQ1 KCTD12 LETM2 PRPH PXMP2 ARNTL2 CDH3 SHC2 SLC20A2 HSPA2 KIAA0182 LIMK2 NAB1 RASIP1 ASRGL1 CLDN3 DCBLD1 SNX10 SSH1 KREMEN2 LIPE NDRG2 ATF3 CLU DCHS1 SOD3 ST3GAL4 MAL LRRC1 NR3C2 ATP8B3 CYC1 DGCR8 EBAG9 SYNGR1 TYMS MCM2 NRG2 RHOF DAGLA DISP2 FAM19A5 TNNI3 UNC5B MYB PAK6 RIPK4 DAZAP1 DOCK3 FBXO6 HSPA4L WHSC1 PNMT PCDH1 B. Hypermethylated-downregulated Genes (n= 31) ARHGAP22 TNFSF9 KLF6 LRP8 NRP1 PAPSS2 SLC43A2 TBC1D16 ASB2 DZIP1 TPM1 MDGA1 NRP2 PIK3CD SMARCA2 TLL2 C18orf1 FBN1 LHFPL2 TRIO NTNG2 PTGIS SOCS2 TNFAIP8 DIXDC1 KIFC3 LMO1 NR3C1 ODZ3 PTPRM SYNPO Supplementary Table 2. Genes enriched for different histone methylation marks in A4 progression model identified through ChIP-on-chip a. H3K4me3 (n= 978) AATF C20orf149 CUL3 FOXP1 KATNA1 NEGR1 RAN SPIN2B ABCA7 C20orf52 CWF19L1 FRK KBTBD10 NEIL1 RANBP2 SPPL2A ABCC9 C21orf13-SH3BGR CXCL3 FSIP1 KBTBD6 NELF RAPGEF3 SPRY4 ABCG2 C21orf45 CYC1 FUK KCMF1 NFKB2 RARB SPRYD3 ABHD7 C22orf32 CYorf15A FXR2 KCNH7 NGDN RASAL2 SPTLC2 ACA15 C2orf18 DAXX FZD9 KCNMB4 NKAP RASD1 SRFBP1 ACA26 C2orf29 DAZ3 G6PD KCTD18 NKTR RASEF SRI ACA3 C2orf32 DBF4 GABPB2 KDELR2 NNT RASGRF1 SRM ACA48 C2orf55 DBF4B GABRA5 KIAA0100 NOL5A RASSF1 SSH2 ACAT1 C3orf44 DBI GADD45B KIAA0226 NOLC1 RASSF3 SSH3 ACSL5 -

Uncovering Genomic Features and Maternal Origin of Korean Native Chicken by Whole Genome Sequencing
RESEARCH ARTICLE Uncovering Genomic Features and Maternal Origin of Korean Native Chicken by Whole Genome Sequencing Woori Kwak1,2.", Ki-Duk Song3.", Jae-Don Oh3, Kang-Nyeong Heo4, Jun-Heon Lee5, Woon Kyu Lee6, Sook Hee Yoon7, Heebal Kim7, Seoae Cho1, Hak-Kyo Lee3* 1. C & K genomics, Seoul, Republic of Korea, 2. Interdisciplinary Program in Bioinformatics, Seoul National University, Seoul, Republic of Korea, 3. Genomic Informatics Center, Hankyong National University, Anseong, Republic of Korea, 4. Poultry Science Division, National Institute of Animal Science, Cheonan, Republic of Korea, 5. Department of Animal Science and Biotechnology, Chungnam National University, Daejeon, Republic of Korea, 6. Inha Research Institute for Medical Sciences, Inha University School of Medicine, Inchon, Republic of Korea, 7. Department of Agricultural Biotechnology and Research Institute for Agriculture and Life Sciences, Seoul National University, Seoul, Republic of Korea OPEN ACCESS *[email protected] Citation: Kwak W, Song K-D, Oh J-D, Heo K-N, Lee J-H, et al. (2014) Uncovering Genomic . These authors contributed equally to this work. Features and Maternal Origin of Korean Native "WK and KDS are co-first authors on this work. Chicken by Whole Genome Sequencing. PLoS ONE 9(12): e114763. doi:10.1371/journal.pone. 0114763 Editor: Axel Janke, BiK-F Biodiversity and Climate Research Center, Germany Abstract Received: March 22, 2014 The Korean Native Chicken (KNC) is an important endemic biological resource in Accepted: November 13, 2014 Korea. While numerous studies have been conducted exploring this breed, none Published: December 11, 2014 have used next-generation sequencing to identify its specific genomic features. -
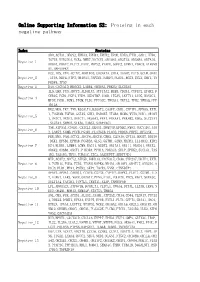
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

Tolerance of Deleterious Mutations on Olfactory Receptors ⇑ Denis Pierron A,B, Nicolás Gutiérrez Cortés B, Thierry Letellier B, Lawrence I
Molecular Phylogenetics and Evolution 66 (2013) 558–564 Contents lists available at SciVerse ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Current relaxation of selection on the human genome: Tolerance of deleterious mutations on olfactory receptors ⇑ Denis Pierron a,b, Nicolás Gutiérrez Cortés b, Thierry Letellier b, Lawrence I. Grossman a, a Center for Molecular Medicine and Genetics, Wayne State University School of Medicine, Detroit, MI 48201, USA b Laboratoire de Physiopathologie Mitochondriale, INSERM, Université Victor Segalen Bordeaux 2, 146, rue Léo Saignat, 33076 Bordeaux, France article info abstract Article history: Knowledge and understanding about the selective pressures that have shaped present human genetic Available online 10 August 2012 diversity have dramatically increased in the last few years in parallel with the availability of large geno- mic datasets. The release of large datasets composed of millions of SNPs across hundreds of genomes by Keywords: HAPMAP, the Human Genome Diversity Panel, and other projects has led to considerable effort to detect Negative selection selection signals across the nuclear genome (Coop et al., 2009; Lopez Herraez et al., 2009; Sabeti et al., Human evolution 2006, 2007; Voight et al., 2006). Most of the research has focused on positive selection forces although Olfactory receptors other selective forces, such as negative selection, may have played a substantive role on the shape of Deleterious SNPs our genome. Here we studied the selective strengths acting presently on the genome by making compu- tational predictions of the pathogenicity of nonsynonymous protein mutations and interpreting the dis- tribution of scores in terms of selection.