Silencing Parasitism Effectors of the Root Lesion Nematode, Pratylenchus Thornei
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Investigation of the Development of Root Lesion Nematodes, Pratylenchus Spp
Türk. entomol. derg., 2021, 45 (1): 23-31 ISSN 1010-6960 DOI: http://dx.doi.org/10.16970/entoted.753614 E-ISSN 2536-491X Original article (Orijinal araştırma) Investigation of the development of root lesion nematodes, Pratylenchus spp. (Tylenchida: Pratylenchidae) in three chickpea cultivars Kök lezyon nematodlarının, Pratylenchus spp. (Tylenchida: Pratylenchidae) üç nohut çeşidinde gelişmesinin incelenmesi İrem AYAZ1 Ece B. KASAPOĞLU ULUDAMAR1* Tohid BEHMAND1 İbrahim Halil ELEKCİOĞLU1 Abstract In this study, penetration, population changes and reproduction rates of root lesion nematodes, Pratylenchus neglectus (Rensch, 1924), Pratylenchus penetrans (Cobb, 1917) and Pratylenchus thornei Sher & Allen, 1953 (Tylenchida: Pratylenchidae), at 3, 7, 14, 21, 28, 35, 42, 49 and 56 d after inoculation in chickpea Bari 2, Bari 3 (Cicer reticulatum Ladiz) and Cermi [Cicer echinospermum P.H.Davis (Fabales: Fabaceae)] were assessed in a controlled environment room in 2018-2019. No juveniles were observed in the roots in the first 3 d after inoculation. Although, population density of P. thornei reached the highest in Cermi (21 d), Bari 3 (42 d) and the lowest observed on Bari 2. Pratylenchus neglectus reached the highest population density in Bari 3 and Cermi on day 28. The population density of P. neglectus was the lowest in Bari 2. Also, population density of P. penetrans reached the highest in Bari 3 cultivar within 49 d, similar to P. thornei, whereas Bari 2 and Cermi had low population densities during the entire experimental period. Keywords: -

Influence of Pratylenchus Vulnus and Meloidogyne Hapla on the Growth of Rootstocks of Rose~ G
influence of Pratylenchus vulnus and Meloidogyne hapla on the Growth of Rootstocks of Rose~ G. S. SANTO 2 and BERT LEAR '~ Abstract: Pratylenchus vulnus is involved in a disease of Rosa noisettiana 'Manetti" rose rootstock characterized by darkening of roots, death of feeder toots, and stunting of entire plants. The disease is more severe when plants are grown in silt loam soil than when they are grown in sandy loam soil. The nematodes reproduce best in silt loam soil at 20 C. Meloidogyne hapla did not affect the growth of Manetti. Rosa sp. 'Dr. Huey', Manetti, and R. odorata rose rootstocks were found to be good hosts for P. vulnus whereas R. multiflora was less suitable. M. hapla re- produced well on R. odorata, Dr. Huey, and R. multi[lora, but not on Maneni. Key Words: root- lesion nematode, root-knot nematode, reproduction, soil temperature, soil type. Pratylenchus vulnus Allen and Jensen roses was associated with a reduction in the was first reported from rose roots in Cali- population of M. hapla and Xiphinema fornia in 1953 (14). A 1970 survey of com- arnericanum Cobb as a result of multiple mercial rose greenhouses in northern Cali- applications of 1,2-dibromo-3-chloropro- fornia shows that this nematode is now pane (DBCP) (8). widely distributed (9), and Allen and Jen- Rosa noisettiana Thory 'Manetti' is the sen (l) also report that P. vulnus is widely most popular rootstock used in growing distributed on field-grown roses in southern greenhouse roses for cut flowers. R. odorata California. Sweet and Rosa sp. -

The Complete Mitochondrial Genome of the Columbia Lance Nematode
Ma et al. Parasites Vectors (2020) 13:321 https://doi.org/10.1186/s13071-020-04187-y Parasites & Vectors RESEARCH Open Access The complete mitochondrial genome of the Columbia lance nematode, Hoplolaimus columbus, a major agricultural pathogen in North America Xinyuan Ma1, Paula Agudelo1, Vincent P. Richards2 and J. Antonio Baeza2,3,4* Abstract Background: The plant-parasitic nematode Hoplolaimus columbus is a pathogen that uses a wide range of hosts and causes substantial yield loss in agricultural felds in North America. This study describes, for the frst time, the complete mitochondrial genome of H. columbus from South Carolina, USA. Methods: The mitogenome of H. columbus was assembled from Illumina 300 bp pair-end reads. It was annotated and compared to other published mitogenomes of plant-parasitic nematodes in the superfamily Tylenchoidea. The phylogenetic relationships between H. columbus and other 6 genera of plant-parasitic nematodes were examined using protein-coding genes (PCGs). Results: The mitogenome of H. columbus is a circular AT-rich DNA molecule 25,228 bp in length. The annotation result comprises 12 PCGs, 2 ribosomal RNA genes, and 19 transfer RNA genes. No atp8 gene was found in the mitog- enome of H. columbus but long non-coding regions were observed in agreement to that reported for other plant- parasitic nematodes. The mitogenomic phylogeny of plant-parasitic nematodes in the superfamily Tylenchoidea agreed with previous molecular phylogenies. Mitochondrial gene synteny in H. columbus was unique but similar to that reported for other closely related species. Conclusions: The mitogenome of H. columbus is unique within the superfamily Tylenchoidea but exhibits similarities in both gene content and synteny to other closely related nematodes. -

Nematodes and Agriculture in Continental Argentina
Fundam. appl. NemalOl., 1997.20 (6), 521-539 Forum article NEMATODES AND AGRICULTURE IN CONTINENTAL ARGENTINA. AN OVERVIEW Marcelo E. DOUCET and Marîa M.A. DE DOUCET Laboratorio de Nematologia, Centra de Zoologia Aplicada, Fant/tad de Cien.cias Exactas, Fisicas y Naturales, Universidad Nacional de Cordoba, Casilla df Correo 122, 5000 C6rdoba, Argentina. Acceplecl for publication 5 November 1996. Summary - In Argentina, soil nematodes constitute a diverse group of invertebrates. This widely distributed group incJudes more than twO hundred currently valid species, among which the plant-parasitic and entomopathogenic nematodes are the most remarkable. The former includes species that cause damages to certain crops (mainly MeloicU:igyne spp, Nacobbus aberrans, Ditylenchus dipsaci, Tylenchulus semipenetrans, and Xiphinema index), the latter inc1udes various species of the Mermithidae family, and also the genera Steinernema and Helerorhabditis. There are few full-time nematologists in the country, and they work on taxonomy, distribution, host-parasite relationships, control, and different aspects of the biology of the major species. Due tO the importance of these organisms and the scarcity of information existing in Argentina about them, nematology can be considered a promising field for basic and applied research. Résumé - Les nématodes et l'agriculture en Argentine. Un aperçu général - Les nématodes du sol représentent en Argentine un groupe très diversifiè. Ayant une vaste répartition géographique, il comprend actuellement plus de deux cents espèces, celles parasitant les plantes et les insectes étant considèrées comme les plus importantes. Les espèces du genre Me/oi dogyne, ainsi que Nacobbus aberrans, Dùylenchus dipsaci, Tylenchulus semipenetrans et Xiphinema index représentent un réel danger pour certaines cultures. -

ENTO-364 (Introducto
K. K. COLLEGE OF AGRICULTURE, NASHIK DEPARTMENT OF AGRICULTURAL ENTOMOLOGY THEORY NOTES Course No.:- ENTO-364 Course Title: - Introductory Nematology Credits: - 2 (1+1) Compiled By Prof. T. B. Ugale & Prof. A. S. Mochi Assistant Professor Department of Agricultural Entomology 0 Complied by Prof. T. B. Ugale & Prof. A. S. Mochi (K. K. Wagh College of Agriculture, Nashik) TEACHING SCHEDULE Semester : VI Course No. : ENTO-364 Course Title : Introductory Nematology Credits : 2(1+1) Lecture Topics Rating No. 1 Introduction- History of phytonematology and economic 4 importance. 2 General characteristics of plant parasitic nematodes. 2 3 Nematode- General morphology and biology. 4 4 Classification of nematode up to family level with 4 emphasis on group of containing economical importance genera (Taxonomic). 5 Classification of nematode by habitat. 2 6 Identification of economically important plant nematodes 4 up to generic level with the help of key and description. 7 Symptoms caused by nematodes with examples. 4 8 Interaction of nematodes with microorganism 4 9 Different methods of nematode management. 4 10 Cultural methods 4 11 Physical methods 2 12 Biological methods 4 13 Chemical methods 2 14 Entomophilic nematodes- Species Biology 2 15 Mode of action 2 16 Mass production techniques for EPN 2 Reference Books: 1) A Text Book of Plant Nematology – K. D. Upadhay & Kusum Dwivedi, Aman Publishing House 2) Fundamentals of Plant Nematology – E. J. Jonathan, S. Kumar, K. Deviranjan, G. Rajendran, Devi Publications, 8, Couvery Nagar, Karumanolapam, Trichirappalli, 620 001. 3) Plant Nematodes - Methodology, Morphology, Systematics, Biology & Ecology Majeebur Rahman Khan, Department of Plant Protection, Faculty of Agricultural Sciences, Aligarh Muslim University, Aligarh, India. -

Phylogenetic Analysis of Nematodes of the Genus Pratylenchus Using Nuclear 26S Rdna
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications from the Harold W. Manter Laboratory of Parasitology Parasitology, Harold W. Manter Laboratory of February 1997 Phylogenetic Analysis of Nematodes of the Genus Pratylenchus Using Nuclear 26S rDNA Luma Al-Banna University of Jordan, [email protected] Valerie M. Williamson University of California, Davis, [email protected] Scott Lyell Gardner University of Nebraska - Lincoln, [email protected] Follow this and additional works at: https://digitalcommons.unl.edu/parasitologyfacpubs Part of the Parasitology Commons Al-Banna, Luma; Williamson, Valerie M.; and Gardner, Scott Lyell, "Phylogenetic Analysis of Nematodes of the Genus Pratylenchus Using Nuclear 26S rDNA" (1997). Faculty Publications from the Harold W. Manter Laboratory of Parasitology. 52. https://digitalcommons.unl.edu/parasitologyfacpubs/52 This Article is brought to you for free and open access by the Parasitology, Harold W. Manter Laboratory of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications from the Harold W. Manter Laboratory of Parasitology by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Published in Molecular Phylogenetics and Evolution (ISSN: 1055-7903), vol. 7, no. 1 (February 1997): 94-102. Article no. FY960381. Copyright 1997, Academic Press. Used by permission. Phylogenetic Analysis of Nematodes of the Genus Pratylenchus Using Nuclear 26S rDNA Luma Al-Banna*, Valerie Williamson*, and Scott Lyell Gardner1 *Department of Nematology, University of California at Davis, Davis, California 95676-8668 1H. W. Manter Laboratory, Division of Parasitology, University of Nebraska State Museum, W-529 Nebraska Hall, University of Nebraska-Lincoln, Lincoln, NE 68588-0514; [email protected] Fax: (402) 472-8949. -

Diversity, Phylogeny, Characterization and Diagnostics of Root-Knot and Lesion Nematodes
Diversity, phylogeny, characterization and diagnostics of root-knot and lesion nematodes Toon Janssen Promotors: Prof. Dr. Wim Bert Prof. Dr. Gerrit Karssen Thesis submitted to obtain the degree of doctor in Sciences, Biology Proefschrift voorgelegd tot het bekomen van de graad van doctor in de Wetenschappen, Biologie 1 Table of contents Acknowledgements Chapter 1: general introduction 1 Organisms under study: plant-parasitic nematodes .................................................... 11 1.1 Pratylenchus: root-lesion nematodes ..................................................................................... 13 1.2 Meloidogyne: root-knot nematodes ....................................................................................... 15 2 Economic importance ..................................................................................................... 17 3 Identification of plant-parasitic nematodes .................................................................. 19 4 Variability in reproduction strategies and genome evolution ..................................... 22 5 Aims .................................................................................................................................. 24 6 Outline of this study ........................................................................................................ 25 Chapter 2: Mitochondrial coding genome analysis of tropical root-knot nematodes (Meloidogyne) supports haplotype based diagnostics and reveals evidence of recent reticulate evolution. 1 Abstract -

A Reappraisal of Tylenchina (Nemata) 1
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications from the Harold W. Manter Laboratory of Parasitology Parasitology, Harold W. Manter Laboratory of 1987 A Reappraisal of Tylenchina (Nemata) 1. For a New Approach to the Taxonomy of Tylenchina Michel Luc Muséun1 national d'Histoire naturelle Armand R. Maggenti University of California - Davis Renaud Fortuner California Department of Food and Agriculture Dewey J. Raski University of California - Davis Etienne Geraert Instituut voor Dierkunde Follow this and additional works at: https://digitalcommons.unl.edu/parasitologyfacpubs Part of the Parasitology Commons Luc, Michel; Maggenti, Armand R.; Fortuner, Renaud; Raski, Dewey J.; and Geraert, Etienne, "A Reappraisal of Tylenchina (Nemata) 1. For a New Approach to the Taxonomy of Tylenchina" (1987). Faculty Publications from the Harold W. Manter Laboratory of Parasitology. 109. https://digitalcommons.unl.edu/parasitologyfacpubs/109 This Article is brought to you for free and open access by the Parasitology, Harold W. Manter Laboratory of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications from the Harold W. Manter Laboratory of Parasitology by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Tribune A REAPPRAISAL OF "IYL,ENCHINA (NEMATA) 1. FOR A NEW APPROACH TO THE TAXONOMY OF TYLENCHINA Michel LUC",Armand R. MAGGENTI**, Renaud FORTUNER***, Dewey J. RASKI** and Etienne GERAERT**** * Muséun1 national d'Histoire naturelle, Laboratoire des Vers, 61, rue de Buffon, 75005 Paris; ** Division of Nematology, University of California, Davis, CA 95616, USA; *** California Department of Food and Agriculture, Analysis and Identification (Nematology), 1220 N. Street, Sacramento, CA 95814, USA, and **** Rijksuniversiteit Gent, Instituut voor Dierkunde, Ledeganckstraat 35, 9000 Gent, Belgium. -

Sudan University of Science and Technology College of Graduate
Sudan University of Science and Technology College of Graduate Studies Studies on Plant Parasitic Nematodes on Banana in Sennar and Kassala States دراﺳﺎت ﻋﻠﻰ اﻟﻨﯿﻤﺎﺗﻮدا اﻟﻤﺘﻄﻔﻠﺔ ﻋﻠﻰ ﻧﺒﺎت اﻟﻤﻮز ﻓﻰ وﻻﯾﺘﻰ ﺳﻨﺎر وﻛﺴﻼ A thesis submitted in to the Sudan University of Science and Technology in Fulfillment of the requirement for M.Sc. (Agric) in crop protection (Plant Pathology) By Duria Abdelsalam Eltahir Elshakh Supervisor: Dr. Ibrahim Saeed Mohammed 2014 ﺑﺳم ﷲ اﻟرﺣﻣن اﻟرﺣﯾم Sudan University of Science and Technology College of Graduate Studies Studies on Plant Parasitic Nematodes on Banana in Sennar and Kassala States دراﺳﺎت ﻋﻠﻰ اﻟﻨﯿﻤﺎﺗﻮدا اﻟﻤﺘﻄﻔﻠﺔ ﻋﻠﻰ ﻧﺒﺎت اﻟﻤﻮز ﻓﻰ وﻻﯾﺘﻰ ﺳﻨﺎر وﻛﺴﻼ A thesis submitted in the requirement of the Degree of M.Sc. Agric. In Crop Protection (Plant Pathology) By Duria Abdelsalam Eltahir Elshakh B.Sc. in Crop Protection University of Zagazig, Egypt(1986) Main Supervisor: Dr. Ibrahim Saeed Mohammed Co. Supervisor: Prof. Gamal Abdalla Elbadri 2014 Dedication To the soul of my father Lovely mother Intimate husband Brothers and sisters To all those who search of knowledge 3 ACKNOWLEDGMENT Thanks and praise to the Beneficent and Merciful God who provided me with health, strength and patience to have this drop from the renewable flood of knowledge. I am gratefully to my supervisors, Doctor Ibrahim Saeed Mohammed and Professor Gamal Abdalla Elbadri for all the encouragement, helpful guidance and continued support. Special thankS to my Co. supervisor Professor. Gamal Abdalla Elbadri under who's the supervision of work has been successfully carried out with his invaluable advices, unlimited helpful and for this correction of this study. -

Functional Diversity of Soil Nematodes in Relation to the Impact of Agriculture—A Review
diversity Review Functional Diversity of Soil Nematodes in Relation to the Impact of Agriculture—A Review Stela Lazarova 1,* , Danny Coyne 2 , Mayra G. Rodríguez 3 , Belkis Peteira 3 and Aurelio Ciancio 4,* 1 Institute of Biodiversity and Ecosystem Research, Bulgarian Academy of Sciences, 2 Y. Gagarin Str., 1113 Sofia, Bulgaria 2 International Institute of Tropical Agriculture (IITA), Kasarani, Nairobi 30772-00100, Kenya; [email protected] 3 National Center for Plant and Animal Health (CENSA), P.O. Box 10, Mayabeque Province, San José de las Lajas 32700, Cuba; [email protected] (M.G.R.); [email protected] (B.P.) 4 Consiglio Nazionale delle Ricerche, Istituto per la Protezione Sostenibile delle Piante, 70126 Bari, Italy * Correspondence: [email protected] (S.L.); [email protected] (A.C.); Tel.: +359-8865-32-609 (S.L.); +39-080-5929-221 (A.C.) Abstract: The analysis of the functional diversity of soil nematodes requires detailed knowledge on theoretical aspects of the biodiversity–ecosystem functioning relationship in natural and managed terrestrial ecosystems. Basic approaches applied are reviewed, focusing on the impact and value of soil nematode diversity in crop production and on the most consistent external drivers affecting their stability. The role of nematode trophic guilds in two intensively cultivated crops are examined in more detail, as representative of agriculture from tropical/subtropical (banana) and temperate (apple) climates. The multiple facets of nematode network analysis, for management of multitrophic interactions and restoration purposes, represent complex tasks that require the integration of different interdisciplinary expertise. Understanding the evolutionary basis of nematode diversity at the field Citation: Lazarova, S.; Coyne, D.; level, and its response to current changes, will help to explain the observed community shifts. -
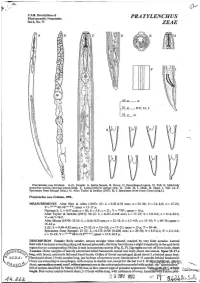
Pratylenchus Zeae
/p C.I.H. Descriptions of Plant-parasitic Nematodes PRATYLENCHUS Set 6, No. 77 ZEAE ' A D F V L 50 P A i5 ,B-F,H,I 25P ,G G Prutylenchus zeue Graham. A-G. Female. A. Entire female. B. Ovary. C. Oesophageal region. D. Tail. E. Mid-body ' transverse section showing lateral fields. F. Lateral field in surface view. G. Tails. H, I. Male. H. Head. I. Tail. (A-F. Specimens from Senegal (orig.). G. After Taylor & Jenkins (1957). H, I. Specimens from Ivory Coast (orig.).) PratyZemhus zeae Graham, 1951. MEASUREMENTS After Sher & Allen (1953): 99: L = 0.36-0.58 mm; a = 25-30; b = 5.4-8.0; c= 17-21; V = 26-43 68-763.4-6."; spear = 15-17 p. Neotype 9: L = 0.47 mm; a = 26; b = 5.9; c = 21 ; V = 30704;spear = 16 p. After Taylor & Jenkins (1957): 90 99: L = 0.413-0.639 mm; a = 17-25; b = 5.0-9.6; c = 11.2-24.1; v = 64.7-74.9. After Memy (1970): 25 99: L = 0.34-0.55 mm; a = 22-33; b = 3.3-4.9; c = 13-18; V = 69-74; spear = 15-18 p. 5 88: L = 0.40-0.42 mm; a = 27-32; b = 3.6-5.0; c = 17-21 ;spear = 15 p; T = 30-44. Specimens from Senegal: 25 99: L = 0.373-0.506 (0.428) mm; a = 20-30; b = 4.9-6.1 ; b' = 3.2-4.6; c = 15-19; V = 23-38 68.6-73.93-86.7; spear= 15.5-16.5 p. -

Vol.41.P.91-99
DISTRIBUTION AND MOLECULAR IDENTIFICATION OF ROOT LESION NEMATODES IN TEMPERATE FRUIT ORCHARDS OF TURKEY Mehmet Ali Söğüt1* and Zübeyir Devran2 1*Süleyman Demirel University, Faculty of Agriculture, Plant Protection Department, 32260 Isparta, TURKEY; 2Batı Akdeniz Agricultural Research Institute (BATEM), Antalya, TURKEY; *Corresponding author: [email protected] ABSTRACT Söğüt, M.A. and Devran, Z., 2011. Distribution and Molecular Identification of Root Lesion Nematodes in Temperate Fruit Orchards of Turkey. Nematropica 41:91-99. Root lesion nematodes are important migratory endoparasitic nematodes attacking temperate fruits in the West Mediterranean region of Turkey. A rapid and accurate method to identify Pratylenchus to the species level is necessary to develop management strategies. Seventy-eight populations of the root lesion nematode were collected from the temperate fruit production region in Turkey, including fruit orchards in Isparta and Antalya provinces. Species-specific primers and rDNA primers were used to identify Pratylenchus spp. Distribution ratios of the sampled root lesion nematode populations were 50%, 45%, 2.5% and 2.5% for P. thornei, P. neglectus, P. penetrans, and P. crenatus, respectively. The present study indicated that P. thornei and P. neglectus were widespread on temperate fruits in the West Mediterranean region of Turkey. Key words: diagnostic, distribution, temperate fruit, PCR, Pratylenchus spp. RESUMEN Söğüt, M.A. and Devran, Z., 2011. Distribución e Identificación Molecular de Pratylenchus en frutales de Turquía. Nematropica 41:91-99. Los nematodos lesionadores son endoparásitos migratorios de importancia que atacan los frutales de clima templado en la region Mediterránea Occidental de Turquía. Se requiere un método confiable y rápido para identificar las species de Pratylenchus que permita desarrollar estrategias de manejo.