Clinical Trial Protocol Doc
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Bitamoazedthesis.Pdf (2.274Mb)
GASTROINTESTINAL ABSORPTION OF HEPARINS A Dissertation Submitted to the College of Graduate Studies and Research in Partial Fulfillments of the Requirements for the Degree of Doctor of Philosophy in the Graduate Program of Veterinary Biomedical Sciences University of Saskatchewan Saskatoon Bita Moazed Copyright Bita Moazed, November 2009. All Rights Reserved PERMISSION TO USE In presenting this dissertation in partial fulfillment of the requirements for a Doctor of Philosophy degree from the University of Saskatchewan, I agree that the Libraries of this University may make it freely available for inspection. I further agree that permission for copying of this thesis/dissertation in any manner, in whole or in part, for scholarly purposes may be granted by Dr. Linda M. Hiebert who supervised my dissertation work or, in her absence, by the chair of the Graduate Program of Veterinary Biomedical Sciences. It is understood that any copying or publication or use of this dissertation or parts thereof for financial gain shall not be allowed without my written permission. It is also understood that due recognition shall be given to me and the University of Saskatchewan in any scholarly use which may be made of any material in this dissertation. Requests for permission to copy or to make other use of other material in this dissertation in whole or part should be addressed to: Chair of the Graduate Program of Veterinary Biomedical Sciences Western College of Veterinary Medicine University of Saskatchewan 52 Campus Drive Saskatoon, Saskatchewan S7N 5B4 Canada i PREFACE This dissertation has been organized as a series of manuscripts that was or will be submitted for publication in scientific journals. -

Review of Anticoagulant Drugs in Paediatric Thromboembolic Disease
18th Expert Committee on the Selection and Use of Essential Medicines (21 to 25 March 2011) Section 12: Cardiovascular medicines 12.5 Antithrombotic medicines Review of Anticoagulant Drugs in Paediatric Thromboembolic Disease September 2010 Prepared by: Fiona Newall Anticoagulation Nurse Manager/ Senior Research Fellow Royal Children’s Hospital/ The University of Melbourne Melbourne, Australia 1 Contents Intent of Review Review of indications for antithrombotic therapy in children Venous thromboembolism Arterial thrombeombolism Anticoagulant Drugs Unfractionated heparin Mechanism of action and pharmacology Dosing and administration Monitoring Adverse events Formulary Summary recommendations Vitamin K antagonists Mechanism of action and pharmacology Dosing and administration Monitoring Adverse events Formulary Summary recommendations Low Molecular Weight Heparins Mechanism of action and pharmacology Dosing and administration Monitoring Adverse events Formulary Summary recommendations ??Novel anticoagulants Summary 2 1. Intent of Review To review the indications for anticoagulant therapies in children. To review the epidemiology of thromboembolic events in children. To review the literature and collate the evidence regarding dosing, administration and monitoring of anticoagulant therapies in childhood. To review the safety of anticoagulant therapies and supervision required To give recommendations for the inclusion of anticoagulants on the WHO Essential Medicines List 2. Review of indications for antithrombotic therapy in children Anticoagulant therapies are given for the prevention or treatment of venous and/or arterial thrombosis. The process of ‘development haemostasis’, whereby the proteins involved in coagulation change quantitatively and qualitatively with age across childhood, essentially protects children against thrombosis(1-5). For this reason, insults such as immobilization are unlikely to trigger the development of thrombotic diseases in children. -

Dabigatran, Rivaroxaban, and Warfarin in the Oldest Adults With
CLINICAL INVESTIGATION Dabigatran, Rivaroxaban, and Warfarin in the Oldest Adults with Atrial Fibrillation in Taiwan Chao-Lun Lai, MD, PhD,*†‡§ Ho-Min Chen, MS,† Min-Tsun Liao, MD,* and Ting-Tse Lin, MD* mortality and cardiovascular mortality than those who OBJECTIVES: To compare the effectiveness and safety of used warfarin. Reduced-dose dabigatran was also associ- reduced-dose dabigatran, reduced-dose rivaroxaban, and warfa- ated with lower risk of intracranial hemorrhage than war- rin in individuals aged 85 and older with atrial fibrillation (AF). farin. J Am AmGeriatr GeriatrSoc Soc66:1567–1574, 2018. 2018. DESIGN: Retrospective cohort study. SETTING: Taiwan National Health Insurance claims database, 20112015. Key words: dabigatran; rivaroxaban; warfarin; effec- PARTICIPANTS: Individuals with AF aged 85 and older tiveness; safety; octogenarian (mean 88.6) with incident use of oral anticoagulants between June 1, 2012 and May 31, 2015 (N54,722; dabi- gatran 110 mg, n51,489; rivaroxaban 15 mg/10 mg, n51,736; warfarin, n51,497). MEASUREMENTS: Clinical outcomes included all-cause death, cardiovascular death, ischemic stroke, acute myocardial he risk of ischemic stroke is 5 times as high in individ- infarction, arterial embolism or thrombosis, intracranial hem- T uals with atrial fibrillation (AF) than in those with- orrhage, and gastrointestinal hemorrhage needing transfusion. out.1 Warfarin, the classic vitamin K antagonist, can reduce 2 Propensity score–matched analysis was performed, and the the risk of ischemic stroke by approximately 60%, but the marginal proportional hazards model was used to estimate the narrow therapeutic window and risk of bleeding complica- relative risk of various clinical outcomes in a matched tions associated with warfarin therapy have led to its being 1 dabigatran-warfarin cohort (n51,180 in each group) and a underused. -
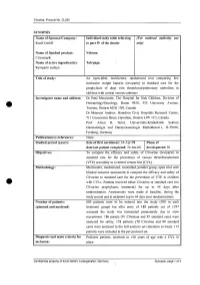
For National Autllority Use Only
Clivarine. Protocol No. CL055 SYNOPSIS Name of Sponsor/Company: Individual study table referring (For national autllOrity use Knoll GmbH to part IV of the dossier only) Name of finished product: Volume: Clivarine® Name of active ingredient(s): Tab/page: Reviparin sodium Title of study: An open-label, multicentre, randomized trial comparing low molecular weight heparin (reviparin) to standard care for the prophylaxis of deep vein thrombosis/pulmonary embolism in children with central venous catheters Investigator name and address: Dr Patti Massicotte, The Hospital for Sick Children, Division of Hematology/Oncology, Room 9420, 555 University Avenue, Toronto, Ontario M5G JX8, Canada Dr Maureen Andrew, Hamilton Civic Hospitals Research Center, 711 Concession Street, Hamilton, Ontario L8V JC3, Canada Prof. Anton H. Sutor, Universitats-Kinderklink Sektion Haematologie und Haemostaseologie Mathildenstr.l, D-79106, Freiburg, Germany Publication(s) (reference): None. Studied period (years): date of first enrolment: 24-Apr-98 Phase of date' last patient completed: 24-Jan-00 development: 111 Objectives: To compare the efficacy and safety of Clivarine (reviparin) to standard care for the prevention of venous thromboembolism (VTE) secondary to a central venous line (CVL). Methodology: Multicentre, randomized, controlled, parallel group, open trial with blinded outcome assessment to compare the efficacy and safety of Clivarine to standard care for the prevention of VTE in children e· with CVLs. Patients received either Clivarine or standard care (no -

DOACS COMPARISON and Faqs
NOACS/DOACS*: PRACTICAL ISSUES AND FREQUENTLY-ASKED QUESTIONS OBJECTIVES: • To summarize characteristics of the direct oral anticoagulants (DOACs) currently available in Canada. • To address common practical issues related to DOAC use. • To address frequently asked questions regarding DOACs. BACKGROUND: The DOACs, which consist of apixaban, dabigatran, edoxaban, and rivaroxaban are used for the prevention and treatment of venous thromboembolism (VTE) and for stroke prevention in atrial fibrillation (AF). Practical advantages of DOACs over warfarin include fixed once- or twice-daily oral dosing without the need for coagulation test monitoring, relatively fewer known drug interactions and no known food interactions. Like warfarin, DOACs increase the risk for bleeding and should be administered under close clinical monitoring. Practical issues regarding the everyday use of DOACs will be addressed in this guide. PRACTICAL AND LIFESTYLE ISSUES: Can DOACs be taken with meals? Rivaroxaban should be taken with meals to enhance absorption; the tablet can be crushed and taken with soft foods such as applesauce. Apixaban, dabigatran, edoxaban can be taken with or without meals. However, taking dabigatran with meals can reduce dyspepsia. Dabigatran capsules should not be opened, broken or chewed before swallowing. Are there any foods or beverages that need to be avoided with DOACs? Unlike with warfarin, there are no known food interactions with DOACs, so there are no food restrictions when taking these medications. In addition, there is no evidence that drinking grapefruit juice affects the effectiveness or safety of DOACs. In general, it is acceptable for patients taking a DOAC to drink alcoholic beverages in moderation (e.g. a glass of wine with a meal) as it is for patients on warfarin. -

Anderson Chapt 8
Chapter-8 Bibliography Where is the evidence? Literature Cited Alpert JS, Dalen JE. Epidemiology and natural history of venous thromboembo- lism. Prog Cardiovas Dis 1994; 36:417-22. Anderson FA, Wheeler HB, Golberg RJ, Hosmer DW, Forcier A, Patwardhan NA. Changing clinical practice: Prospective study of the impact of continuing medical education and quality assurance programs on use of prophylaxis for venous thromboembolism Arch Intern Med 1994; 154:669-677. Anderson FA Jr, Wheeler HB, Goldberg RJ, Hosmer DW, Forcier A, Patwardhan NA. Physician practices in the prevention of venous thromboembolism. Ann Intern Med 1991;115:591-595. Anderson FA Jr, Wheeler HB. Strategies to improve implementation. In: Goldhaber S, ed. Prevention of Venous Thromboembolism. New York, NY: Marcel Dekker Inc.; 1992; 519-539. Bergqvist D. 1983. Post-operative Thromboembolism, Frequency, Etiology, Prophylaxis. Berlin: Springer-Verlag. Clagett GP, Anderson FA Jr, Heit J, Levine MN, Wheeler HB. Prevention of venous thromboembolism. Chest 1995; 108:312S-334S. Collins R, Scrimgeour A, Yusuf S, et al. Reduction in fatal pulmonary embolism and venous thrombosis by perioperative administration of subcutaneous heparin. N Engl J Med 1988; 318:1162-73. Gallus AS. Anticoagulants in the prevention of venous thromboembolism. Baillieres Clin Haematol 1990; 3(3):651-684. Hull RD, Raskob GE, Hirsh J. The diagnosis of clinically suspected pulmonary embolism: Practical approaches. Chest 1986; 89(5 Suppl):417S-425S. Morrell MP, Dunhill MA. The postmortem incidence of pulmonary embolism in a hospital population. Br J Surg 1968; 55:347-352. NIH Consensus Development. Prevention of venous thrombosis and pulmonary Best embolism. JAMA 1986; 256:744-749. -

Atrial Fibrillation
CONFIDENTIAL NATIONAL INSTITUTE FOR HEALTH AND CLINICAL EXCELLENCE Premeeting briefing Dabigatran for the prevention of stroke and systemic embolism in atrial fibrillation This briefing presents the key issues arising from the manufacturer’s submission, Evidence Review Group (ERG) report and statements made by consultees and their nominated clinical specialists and patient experts. Please note that this briefing is a summary of the information available and should be read with the full supporting documents. The manufacturer was asked to…. provide justification for reducing the dabigatran treatment dose from 150 mg to 110 mg at 80 years of age comment on the impact of being unable to utilise P-glycoprotein inhibitors on the use of dabigatran and the management of atrial fibrillation provide a justification for choosing the mixed treatment comparison (MTC) (SAS) for the base case instead of the results from the MTC (WinBUGs) provide a comparison of the different hazard ratios from the MTC (SAS), MTC (WinBUGS) analyses and the direct pairwise results and justify any discrepancies justify the exclusion of trials with zero event arms from the MTC provide a revised model with the ability to choose any of the included treatments (dabigatran or warfarin) as either a first-line or a second-line treatment option provide base-case cost-effectiveness results comparing dabigatran 110 mg and 150 mg when used as either first-line treatment or as a second-line treatment following warfarin analyse and provide base-case cost-effectiveness comparing the results of the following treatment sequences: dabigatran → warfarin → aspirin → no treatment and warfarin → aspirin → no treatment. -

Laboratory Monitoring of Direct Oral Anticoagulants (Doacs)
biomedicines Review Laboratory Monitoring of Direct Oral Anticoagulants (DOACs) Claire Dunois HYPHEN BioMed, Sysmex Group, 95000 Neuville sur Oise, France; [email protected] Abstract: The introduction of direct oral anticoagulants (DOACs), such as dabigatran, rivaroxaban, apixaban, edoxaban, and betrixaban, provides safe and effective alternative to previous anticoagulant therapies. DOACs directly, selectively, and reversibly inhibit factors IIa or Xa. The coagulation effect follows the plasma concentration–time profile of the respective anticoagulant. The short half-life of a DOAC constrains the daily oral intake. Because DOACs have predictable pharmacokinetic and pharmacodynamic responses at a fixed dose, they do not require monitoring. However in specific clinical situations and for particular patient populations, testing may be helpful for patient management. The effect of DOACs on the screening coagulation assays such as prothrombin time (PT), activated partial thromboplastin time (APTT), and thrombin time (TT) is directly linked to reagent composition, and clotting time can be different from reagent to reagent, depending on the DOAC’s reagent sensitivity. Liquid chromatography–mass spectrometry (LC-MS/MS) is considered the gold standard method for DOAC measurement, but it is time consuming and requires expensive equipment. The general consensus for the assessment of a DOAC is clotting or chromogenic assays using specific standard calibrators and controls. This review provides a short summary of DOAC properties and an update on laboratory methods for measuring DOACs. Keywords: DOAC; monitoring; screening assays; quantitative assays Citation: Dunois, C. Laboratory Monitoring of Direct Oral Anticoagulants (DOACs). 1. Introduction Biomedicines 2021, 9, 445. https:// Direct oral anticoagulants (DOACs) constitute first-line therapy used for many throm- doi.org/10.3390/biomedicines9050445 boembolic indications, such as prevention and treatment of venous thromboembolism (VTE) and stroke prevention in atrial fibrillation (AF) [1,2]. -

Anticoagulant Pradaxa (Dabigatran Etexilate Mesylate) Savaysa (Edoxaban) Xarelto 2.5Mg (Rivaroxaban) Effective 01/01/2021
Anticoagulant Pradaxa (dabigatran etexilate mesylate) Savaysa (edoxaban) Xarelto 2.5mg (rivaroxaban) Effective 01/01/2021 ☒ MassHealth Plan ☒ ☐Commercial/Exchange Prior Authorization Program Type ☒ Quantity Limit ☒ Pharmacy Benefit Benefit ☐ Step Therapy ☐ Medical Benefit (NLX) Specialty N/A Limitations Specialty Medications All Plans Phone: 866-814-5506 Fax: 866-249-6155 Non-Specialty Medications Contact MassHealth Phone: 877-433-7643 Fax: 866-255-7569 Information Commercial Phone: 800-294-5979 Fax: 888-836-0730 Exchange Phone: 855-582-2022 Fax: 855-245-2134 Medical Specialty Medications (NLX) All Plans Phone: 844-345-2803 Fax: 844-851-0882 Exceptions N/A Overview Xarelto and Savaysa are factor Xa inhibitors which inhibit platelet activation and fibrin clot formation. Pradaxa is a thrombin inhibitor which blocks free and fibrin bound thrombin. These medications are indicated for: . Treatment of deep venous thrombosis (DVT) and pulmonary embolism (PE) – Pradaxa and Savaysa . Prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. – Pradaxa, Xarelto, and Savaysa . Prophylaxis of DVT and/or PE in patients who have undergone total hip arthroplasty. - Xarelto and Pradaxa . Prophylaxis of venous thromboembolism (VTE) – Xarelto . Reduction in the risk of recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) – Xarelto . Reduction of risk of major cardiovascular (CV) events (CV death, myocardial infarction, and stroke) in patients with coronary artery disease (chronic) or peripheral artery disease. - Xarelto No PA PA required Pradaxa® (dabigatran etexilate mesylate 110 mg) ≤ 70 Pradaxa® (dabigatran etexilate mesylate 75 mg, 150 capsules/365 days mg) Pradaxa® (dabigatran etexilate mesylate 110 mg) > 70 Eliquis® (apixaban) PD capsules/365 days ® ® Xarelto (rivaroxaban 10 mg, 15 mg, 20 mg, starter pack) Savaysa (edoxaban) ® Xarelto (rivaroxaban 2.5 mg tablet) PD Preferred Drug. -

Estonian Statistics on Medicines 2016 1/41
Estonian Statistics on Medicines 2016 ATC code ATC group / Active substance (rout of admin.) Quantity sold Unit DDD Unit DDD/1000/ day A ALIMENTARY TRACT AND METABOLISM 167,8985 A01 STOMATOLOGICAL PREPARATIONS 0,0738 A01A STOMATOLOGICAL PREPARATIONS 0,0738 A01AB Antiinfectives and antiseptics for local oral treatment 0,0738 A01AB09 Miconazole (O) 7088 g 0,2 g 0,0738 A01AB12 Hexetidine (O) 1951200 ml A01AB81 Neomycin+ Benzocaine (dental) 30200 pieces A01AB82 Demeclocycline+ Triamcinolone (dental) 680 g A01AC Corticosteroids for local oral treatment A01AC81 Dexamethasone+ Thymol (dental) 3094 ml A01AD Other agents for local oral treatment A01AD80 Lidocaine+ Cetylpyridinium chloride (gingival) 227150 g A01AD81 Lidocaine+ Cetrimide (O) 30900 g A01AD82 Choline salicylate (O) 864720 pieces A01AD83 Lidocaine+ Chamomille extract (O) 370080 g A01AD90 Lidocaine+ Paraformaldehyde (dental) 405 g A02 DRUGS FOR ACID RELATED DISORDERS 47,1312 A02A ANTACIDS 1,0133 Combinations and complexes of aluminium, calcium and A02AD 1,0133 magnesium compounds A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 811120 pieces 10 pieces 0,1689 A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 3101974 ml 50 ml 0,1292 A02AD83 Calcium carbonate+ Magnesium carbonate (O) 3434232 pieces 10 pieces 0,7152 DRUGS FOR PEPTIC ULCER AND GASTRO- A02B 46,1179 OESOPHAGEAL REFLUX DISEASE (GORD) A02BA H2-receptor antagonists 2,3855 A02BA02 Ranitidine (O) 340327,5 g 0,3 g 2,3624 A02BA02 Ranitidine (P) 3318,25 g 0,3 g 0,0230 A02BC Proton pump inhibitors 43,7324 A02BC01 Omeprazole -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Table of Contents
Table of contents PRISMA checklist .............................................................................................................................................. 2 Tables Supplementary Table S1. Search strategy ..................................................................................................... 4 Supplementary Table S2. Characteristics of included randomized trials, stratified by patient type . 6 Supplementary Table S3. Ongoing trials ..................................................................................................... 10 Supplementary Table S4. Risk of bias assessment ................................... Error! Bookmark not defined. Supplementary Table S5. Sensitivity analysis: best-worse and worst-best case scenario’s ................ 11 Supplementary Table S6. Sensitivity analysis: trials with publication year ≥ 2005 ............................. 16 Supplementary Table S7. Sensitivity analysis: including LMWH types not a priori defined ..... Error! Bookmark not defined. Supplementary Table S8. Subgroup analysis: co-primary outcomes ..................................................... 16 Supplementary Table S9. Sensitivity analysis: proportion of SAE and cumulative SAE ................... 13 Figures Supplementary Figure S1. Forest plot of SAE, stratified for patient type ............................................. 17 Supplementary Figure S2. Forest plot of non-major bleeding, stratified for patient type .................. 18 Supplementary Figure S3. Forest plot of any VTE, stratified