Platelet Aggregometry Testing: Molecular Mechanisms, Techniques and Clinical Implications
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

AA Metabolism, 139 A2b1, 69, 70, 318–323 ABCB1 Gene, 176 Abciximab
Index A aIIbb3, 60, 62, 69, 70, 73, 74, 79, 80 AA. See Arachidonic acid (AA) AJW202, 293 AA metabolism, 139 Akt-1, 345 a2b1, 69, 70, 318–323 Akt-2, 345 ABCB1 gene, 176 ALX-0081, 296 Abciximab, 206–208, 497, 500, 501, 504–505, ALX-0681, 298 509, 513, 514, 525 Anagrelide, 227, 229 approval, 206 Ankle brachial indexes (ABIs), 549, 587, EPIC trial, 207 595–597 EPILOG study, 207 framingham risk score, 551 EPISTENT study, 207 screening, 550 platelet binding, 207 sensitivity, 551 thrombocytopenia, 206 specificity, 551 in unstable angina, 208 vascular risk, 550 ABIs. See Ankle brachial indexes (ABIs) Antagomirs, 439 Acetylsalicylic acid (aspirin), 137–158 Antibodies, 317 ACS. See Acute coronary syndromes (ACS) Anticoagulants, 525, 537 Acute coronary syndromes (ACS), 288 Anticoagulation, 533, 534 Acute ischemic stroke, 143, 149–150 Antioxidant, 233 Acute myocardial infarction, 142, 143, 149, Antiplatelet agents, 263, 553 153, 157 Antithrombotic Trialist’s ADAMTS-13, 93 Collaboration, 553 Adenosine diphosphate (ADP), 37, 88, 96, aspirin, 553 166, 473 clopidogrel, 553 Adenosine triphosphate (ATP), 37 dipyridamole, 553 Adhesion, 90, 112–118, 125 meta-analysis, 560 Adhesion molecules infiltration, 264 picotamide, 553 Adiponectin, 315 risk reduction, 553 ADP. See Adenosine diphosphate (ADP) ticlopidine, 553 ADP inhibitors (or P2Y12 blockers), 497–499, vascular events, 553 501–505, 507–509, 513, 514 Antiplatelet therapy, 472 ADP-receptor antagonists, 169 Apixaban, 535, 539 Adverse effects, 153–156 Aptamers, 299–301 Aegyptin, 327 Arachidonic acid (AA), 474 P. Gresele et al. (eds.), Antiplatelet Agents, Handbook of Experimental 607 Pharmacology 210, DOI 10.1007/978-3-642-29423-5, # Springer-Verlag Berlin Heidelberg 2012 608 Index ARC1779, 299–300 TXS signaling, 279 ARC15105, 300 Cardiovascular death, 248 AR-C69931MX, 456 Carotid endarterectomy (ACE) inhibition, Aspirin, 474, 496, 498–499, 501–502, 143, 156 504–507, 511–514, 520, 522, Carotid stenosis, 523 527, 531, 533–537, 539, CCBs. -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -

Laboratory Testing for Von Willebrand Disease
75 Laboratory Testing for von Willebrand Disease: The Past, Present, and Future State of Play for von Willebrand Factor Assays that Measure Platelet Binding Activity, with or without Ristocetin Sarah Just, B App Sc. MLS, MAIMS1 1 Department of Haematology, South Eastern Area Laboratory Service Address for correspondence SarahJust,BAppSc.MLS,MAIMS, (SEALS), Prince of Wales Hospital, Sydney, New South Wales, Department of Haematology, South Eastern Area Laboratory Service Australia (SEALS), Prince of Wales Hospital, Sydney, NSW 2031, Australia (e-mail: [email protected]). Semin Thromb Hemost 2017;43:75–91. Abstract von Willebrand disease (VWD) was first described nearly a century ago in 1924 by Erik Adolf von Willebrand. Diagnostic testing at the time was very limited and it was not until the mid to late 1900s that more tests became available to assist with the diagnosis and Keywords classification of VWD. Two of these tests are based on ristocetin, one being ristocetin- ► ristocetin cofactor induced platelet aggregation (RIPA) and the other the von Willebrand factor (VWF) ► ristocetin induced ristocetin cofactor assay (VWF:RCo). The VWF:RCo assay provides functional assessment platelet aggregation of in vitro VWF binding to the platelet glycoprotein (Gp) complex, GPIb-IX-V. Despite ► von Willebrand some advancements and newer technologies utilizing the principles of the original disease VWF:RCo assay, the original assay is still referred to as the gold standard for measure- ► laboratory testing ment of VWF activity. This article will review the history of VWD diagnostic assays, ► von Willebrand factor including RIPA and VWF:RCo over the past 40 years, as well as the newer assays that ► von Willebrand factor measure platelet binding with or without ristocetin, and which have been developed activity with the aim to potentially replace platelet-based ristocetin-dependent assays. -
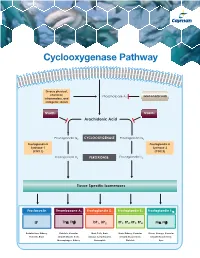
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Effect of Prostanoids on Human Platelet Function: an Overview
International Journal of Molecular Sciences Review Effect of Prostanoids on Human Platelet Function: An Overview Steffen Braune, Jan-Heiner Küpper and Friedrich Jung * Institute of Biotechnology, Molecular Cell Biology, Brandenburg University of Technology, 01968 Senftenberg, Germany; steff[email protected] (S.B.); [email protected] (J.-H.K.) * Correspondence: [email protected] Received: 23 October 2020; Accepted: 23 November 2020; Published: 27 November 2020 Abstract: Prostanoids are bioactive lipid mediators and take part in many physiological and pathophysiological processes in practically every organ, tissue and cell, including the vascular, renal, gastrointestinal and reproductive systems. In this review, we focus on their influence on platelets, which are key elements in thrombosis and hemostasis. The function of platelets is influenced by mediators in the blood and the vascular wall. Activated platelets aggregate and release bioactive substances, thereby activating further neighbored platelets, which finally can lead to the formation of thrombi. Prostanoids regulate the function of blood platelets by both activating or inhibiting and so are involved in hemostasis. Each prostanoid has a unique activity profile and, thus, a specific profile of action. This article reviews the effects of the following prostanoids: prostaglandin-D2 (PGD2), prostaglandin-E1, -E2 and E3 (PGE1, PGE2, PGE3), prostaglandin F2α (PGF2α), prostacyclin (PGI2) and thromboxane-A2 (TXA2) on platelet activation and aggregation via their respective receptors. Keywords: prostacyclin; thromboxane; prostaglandin; platelets 1. Introduction Hemostasis is a complex process that requires the interplay of multiple physiological pathways. Cellular and molecular mechanisms interact to stop bleedings of injured blood vessels or to seal denuded sub-endothelium with localized clot formation (Figure1). -

Diagnosis of Hemophilia and Other Bleeding Disorders
Diagnosis of Hemophilia and Other Bleeding Disorders A LABORATORY MANUAL Second Edition Steve Kitchen Angus McCraw Marión Echenagucia Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2010 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. For permission to reproduce or translate this document, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s website at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel.: (514) 875-7944 Fax: (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org Diagnosis of Hemophilia and Other Bleeding Disorders A LABORATORY MANUAL Second Edition (2010) Steve Kitchen Angus McCraw Marión Echenagucia WFH Laboratory WFH Laboratory (co-author, Automation) Training Specialist Training Specialist Banco Municipal Sheffield Haemophilia Katharine Dormandy de Sangre del D.C. and Thrombosis Centre Haemophilia Centre Universidad Central Royal Hallamshire and Thrombosis Unit de Venezuela Hospital The Royal Free Hospital Caracas, Venezuela Sheffield, U.K. London, U.K. on behalf of The WFH Laboratory Sciences Committee Chair (2010): Steve Kitchen, Sheffield, U.K. Deputy Chair: Sukesh Nair, Vellore, India This edition was reviewed by the following, who at the time of writing were members of the World Federation of Hemophilia Laboratory Sciences Committee: Mansoor Ahmed Clarence Lam Norma de Bosch Sukesh Nair Ampaiwan Chuansumrit Alison Street Marión Echenagucia Alok Srivastava Andreas Hillarp Some sections were also reviewed by members of the World Federation of Hemophilia von Willebrand Disease and Rare Bleeding Disorders Committee. -

2374 Supplementary Drugs and Other Substances
2374 Supplementary Drugs and Other Substances 5. Burdock GA. Review of the biological properties and toxicity of Pharmacokinetics reduced to the endoperoxide prostaglandin H2 (PGH2). Prostag- bee propolis (propolis). Food Chem Toxicol 1998; 36: 347–63. Propylene glycol is rapidly absorbed from the gastrointestinal landin H2 is then converted to the primary prostaglandins pros- 6. Lieberman HD, et al. Allergic contact dermatitis to propolis in a tract. There is evidence of topical absorption when applied to taglandin D2, prostaglandin E2, and prostaglandin F2 , to throm- violin maker. J Am Acad Dermatol 2002; 46 (suppl): S30–S31. damaged skin. boxane A2 (TXA2) via the enzyme thromboxane synthetase, or 7. Giusti F, et al. Sensitization to propolis in 1255 children under- It is extensively metabolised in the liver primarily by oxidation to prostacyclin (PGI2) via the enzyme prostacyclin synthetase. going patch testing. Contact Dermatitis 2004; 51: 255–8. to lactic and pyruvic acid and is also excreted in the urine These products are further metabolised and rapidly inactivated in 8. Walgrave SE, et al. Allergic contact dermatitis from propolis. unchanged. the body. Dermatitis 2005; 16: 209–15. The secondary prostaglandins, prostaglandin A (PGA ), pros- 9. Majiene D, et al. Antifungal and antibacterial activity of propo- ◊ References. 2 2 lis. Curr Nutr Food Sci 2007; 3: 304–8. 1. Yu DK, et al. Pharmacokinetics of propylene glycol in humans taglandin B2 (PGB2), and prostaglandin C2 (PGC2) are derived during multiple dosing regimens. J Pharm Sci 1985; 74: 876–9. from prostaglandin E2, but are formed during extraction and Preparations 2. Speth PAJ, et al. -

Platelet Function Disorders
TREATMENT OF HEMOPHILIA APRIL 2008 • NO 19 PLATELET FUNCTION DISORDERS Second Edition Anjali A. Sharathkumar Amy Shapiro Indiana Hemophilia and Thrombosis Center Indianapolis, U.S.A. Published by the World Federation of Hemophilia (WFH), 1999; revised 2008. © World Federation of Hemophilia, 2008 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s website at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff. -

COX-1 and COX-2 Enzymes Synthesize Prostaglandins and Are Teacher Emeritus, University of Wisconsin-Madison) Mentor: Dr
COX-1 And COX-2 Enzymes Synthesize Prostaglandins and Are Teacher Emeritus, University of Wisconsin-Madison) Mentor: Dr. David Nelson (Professor of Biochemistry, University of Wisconsin- (Student, University of Wisconsin-Madison) Center for Inhibited by NSAIDS (Nonsteroidal Anti-inflammatory Drugs) BioMolecular Madison West High School: Audra Amasino, Yuting Deng, Samuel Huang, Iris Lee, Adeyinka Lesi, Yaoli Pu, and Peter Vander Velden Modeling Advisor: Gary Graper, Teacher Emeritus, University of Wisconsin-Madison Mentors: Dr. David Nelson, Professor of Biochemistry, and Basudeb Bhattacharyya, Student, University of Wisconsin-Madison Abstract Prostaglandin Hormone Synthases (COX-1 and COX-2) are enzymes that produce prostaglandins. Prostaglandins are (1) Structure (3) Cyclooxygenase Active Sites responsible for fever, pain, and inflammation, but also the (5) Drugs maintenance of the lining of the stomach and prevention of In the pictures below, the heme is orange, the ulceration. COX-1 is found mainly in the gastrointestinal lining, hydrophobic knob is yellow, and the amino acids in the The three drugs below are all nonsteroidal anti- and COX-2 at sites of inflammation. NSAIDS (Nonsteroidal anti- Cyclooxygenase active site are colored in CPK (red for inflammatory drugs (NSAIDS). The first two NSAIDS, inflammatory drugs) such as aspirin, ibuprofen, naproxen, and oxygen, blue for nitrogen, and gray for carbon). aspirin and ibuprofen, are called nonselective Cox flurbiprofen inhibit both COX-1 and COX-2, and are taken inhibitors since they affect both COX-1 and COX-2 regularly by over 33 million Americans for pain and substantially (note their high COX-2/COX-1 effect inflammation. Some 10%-50% of these users suffer ratios). -

Effects of Prostaglandin H2 on Perinatal Pulmonary Circulation
003 1-3998/86/2006-0565$02.00/0 PEDIATRIC RESEARCH Vol. 20, No. 6, 1986 Copyright O 1986 International Pediatric Research Foundation, Inc. Printed in U.S.A. Effects of Prostaglandin H2 on Perinatal Pulmonary Circulation MARY L. TOD,' SIDNEY CASSIN, DENNIS B. McNAMARA, AND PHILIP J. KADOWITZ Departments of Physiology and Pediatrics, College of Medicine, University of Florida, Gainesville, Florida 32610 and Department of Pharmacology, Tulane Medical School, New Orleans, Louisiana 70112 ABSTRACT. A pivotal intermediate in prostaglandin (PG) terminal enzymes that yield these various products are present biosynthesis is the endoperoxide PGH2. This endoperox- in varying amounts and activities in different organs, and this ide is capable of eliciting direct responses in biological distribution contributes to the wide range of responses of indi- systems without undergoing conversion to other PGs. Ef- vidual systems to release of PGs (1-6). There is evidence that fects of PGH2 include stimulation of platelet aggregation PGH2 is capable of eliciting direct responses to biological systems and vascular smooth muscle contraction in vitro; injections without undergoing conversion to other PGs (7, 8). Kadowitz et of PGH2 in vivo cause increases in pulmonary arterial al. (9) demonstrated the ability of PGH2 to contract isolated pressure. The response of the pulmonary vasculature of segments of canine intrapulmonary vein. Additionally, bolus perinatal lambs to PGH2 was measured using an in situ injections of PGH2 caused increases in lobar arterial and small pump-perfused left lower lung preparation. Intrapulmonary vein pressures in pump-perfused canine lungs (9). Unanes- injections of PGH2 (0.24-0.61 pg/kg) into six unventilated thetized adult sheep responded to infusions of PGH2 with dose- fetal lambs (0.93-0.97 gestation) produced decreases in related increases of pulmonary arterial pressure (10). -

Isolation and Structure of Two Prostaglandin Endoperoxides That
Proc. Nat. Acad. Sci. USA Vol. 71, No. 2, pp. 345-349, February 1974 Isolation and Structure of Two Prostaglandin Endoperoxides That Cause Platelet Aggregation (15-hydroperoxy endoperoxide/15-hydroxy endoperoxide/platelet aggregation/ contraction of rabbit aorta) MATS HAMBERG, JAN SVENSSON, TOSHIO WAKABAYASHI, AND BENGT SAMUELSSON Department of Chemistry, Karolinska Institutet, S 104 01, Stockholm, Sweden Communicated by Hugo Theorell, September 19, 1978 ABSTRACT Incubation for a short time of arachidonic (kindly provided by Dr. W. Stoffel, Cologne, Germany, see acid with the microsomal fraction of a homogenate of the ref. 2) and Na14CN followed by hydrolysis of the nitrile. The vesicular gland of sheep in the presence of 1 mM p-mer- curibenzoate followed by extraction and silicic acid chemical and radiochemical purity was in excess of 98%, chromatography yielded two prostaglandin endoper- as judged by thin-layer radiochromatography. Part of the oxides. The structures of these compounds, i.e., 15-hy- labeled acid was diluted with unlabeled material to make a droperoxy-9a,11a-peroxidoprosta-5,13-dienoic acid (pros- preparation with specific radioactivity of 0.77 Ci/mol, which taglandin G2) and 15-hydroxy-9a,lla-peroxidoprosta- 5,13-dienoic acid (prostaglandin H2), were assigned mainly was used for incubations with vesicular gland microsomes. by a number of chemical transformations into previously Thin-Layer Chromatography (TLC) was carried out with known prostaglandins. The new prostaglandins were 50- 200 times (prostaglandin G2) and 100-450 times (prosta- plates coated with chloroform-methanol-washed Silica gel G glandin H2) more active than prostaglandin E2 on the super- and the following solvent systems (when not otherwise fused aorta strip. -

Prostacyclin: an Inflammatory Paradox
REVIEW ARTICLE published: 13 May 2011 doi: 10.3389/fphar.2011.00024 Prostacyclin: an inflammatory paradox Jeremiah Stitham, Charles Midgett, Kathleen A. Martin and John Hwa* Section of Cardiovascular Medicine, Department of Internal Medicine, Yale School of Medicine, Yale University, New Haven, CT, USA Edited by: Prostacyclin (PGI2) is a member of the prostaglandin family of bioactive lipids. Its best- Angel Lanas, University of Zaragoza, characterized role is in the cardiovascular system, where it is released by vascular endothelial cells, Spain serving as a potent vasodilator and inhibitor of platelet aggregation. In recent years, prostacyclin Reviewed by: Emer Smyth, University of (PGI2) has also been shown to promote differentiation and inhibit proliferation in vascular smooth Pennsylvania, USA muscle cells. In addition to these well-described homeostatic roles within the cardiovascular Steven W. Kerrigan, Royal College of system, prostacyclin (PGI2) also plays an important role as an inflammatory mediator. In this Surgeons in Ireland, Ireland review, we focus on the contribution of prostacyclin (PGI2) as both a pathophysiological mediator *Correspondence: and therapeutic agent in three major inflammatory-mediated disease processes, namely John Hwa, Section of Cardiovascular Medicine, Department of Internal rheumatoid arthritis, where it promotes disease progression (“pro-inflammatory”), along Medicine, Yale School of Medicine, with pulmonary vascular disease and atherosclerosis, where it inhibits disease progression Cardiovascular Research Center, 300 (“anti-inflammatory”). The emerging role of prostacyclin (PGI2) in this context provides new George Street, Room 759H, New opportunities for understanding the complex molecular basis for inflammatory-related diseases, Haven, CT 06511, USA. e-mail: [email protected] and insights into the development of current and future anti-inflammatory treatments.