Iloprost Suppresses Connective Tissue Growth Factor Production in Fibroblasts and in the Skin of Scleroderma Patients
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

VTE) Cindy Ward, DNP, RN-BC, CMSRN, ACNS-BC
Chronic Complications of Venous Thromboembolism (VTE) Cindy Ward, DNP, RN-BC, CMSRN, ACNS-BC Roanoke, VA Disclosure • The speaker has no conflicts of interest to disclose. Carilion Roanoke Memorial Hospital • 3 time Magnet® designated • Flagship of Carilion Clinic, a 7 hospital system • Region’s only Level 1 Trauma Center • 703-bed academic medical center • System-wide, serves nearly 1 million patients across Virginia, West Virginia and North Carolina Objectives At the conclusion of this education activity, the learner will be able to: • recall VTE risk factors and prevention • describe post-thrombotic syndrome and chronic thromboembolic pulmonary hypertension (CTEPH) • identify nursing implications of caring for patients with post-thrombotic syndrome and CTEPH What is VTE? Pulmonary Embolus Deep Vein (PE) Thrombosis (DVT) Signs/Symptoms of DVT • Swelling • Erythema • Pain • Warmth1 Signs/Symptoms of PE 1 • Sudden onset of dyspnea • Tachycardia • Irregular heartbeat • Chest pain, worse with deep breath • Hemoptysis • Low blood pressure, light-headedness or syncope • 350,000 – 900,000 people per year are affected by VTE1, 2 • VTE is the leading cause of preventable hospital death in the US2 • VTE can occur without symptoms (silent)3 Quick Facts • Patients with DVT who are untreated have a 37% incidence of PE that is fatal4 • Combined mortality from PE (initial and recurrence) is 73%4 Quick Facts • 1 in 20 hospitalized patients will suffer a fatal PE if they have not received adequate VTE prophylaxis5 • For 25% of patients with PE, the first -

AA Metabolism, 139 A2b1, 69, 70, 318–323 ABCB1 Gene, 176 Abciximab
Index A aIIbb3, 60, 62, 69, 70, 73, 74, 79, 80 AA. See Arachidonic acid (AA) AJW202, 293 AA metabolism, 139 Akt-1, 345 a2b1, 69, 70, 318–323 Akt-2, 345 ABCB1 gene, 176 ALX-0081, 296 Abciximab, 206–208, 497, 500, 501, 504–505, ALX-0681, 298 509, 513, 514, 525 Anagrelide, 227, 229 approval, 206 Ankle brachial indexes (ABIs), 549, 587, EPIC trial, 207 595–597 EPILOG study, 207 framingham risk score, 551 EPISTENT study, 207 screening, 550 platelet binding, 207 sensitivity, 551 thrombocytopenia, 206 specificity, 551 in unstable angina, 208 vascular risk, 550 ABIs. See Ankle brachial indexes (ABIs) Antagomirs, 439 Acetylsalicylic acid (aspirin), 137–158 Antibodies, 317 ACS. See Acute coronary syndromes (ACS) Anticoagulants, 525, 537 Acute coronary syndromes (ACS), 288 Anticoagulation, 533, 534 Acute ischemic stroke, 143, 149–150 Antioxidant, 233 Acute myocardial infarction, 142, 143, 149, Antiplatelet agents, 263, 553 153, 157 Antithrombotic Trialist’s ADAMTS-13, 93 Collaboration, 553 Adenosine diphosphate (ADP), 37, 88, 96, aspirin, 553 166, 473 clopidogrel, 553 Adenosine triphosphate (ATP), 37 dipyridamole, 553 Adhesion, 90, 112–118, 125 meta-analysis, 560 Adhesion molecules infiltration, 264 picotamide, 553 Adiponectin, 315 risk reduction, 553 ADP. See Adenosine diphosphate (ADP) ticlopidine, 553 ADP inhibitors (or P2Y12 blockers), 497–499, vascular events, 553 501–505, 507–509, 513, 514 Antiplatelet therapy, 472 ADP-receptor antagonists, 169 Apixaban, 535, 539 Adverse effects, 153–156 Aptamers, 299–301 Aegyptin, 327 Arachidonic acid (AA), 474 P. Gresele et al. (eds.), Antiplatelet Agents, Handbook of Experimental 607 Pharmacology 210, DOI 10.1007/978-3-642-29423-5, # Springer-Verlag Berlin Heidelberg 2012 608 Index ARC1779, 299–300 TXS signaling, 279 ARC15105, 300 Cardiovascular death, 248 AR-C69931MX, 456 Carotid endarterectomy (ACE) inhibition, Aspirin, 474, 496, 498–499, 501–502, 143, 156 504–507, 511–514, 520, 522, Carotid stenosis, 523 527, 531, 533–537, 539, CCBs. -

CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene and Pathophysiological Conditions
Review CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene and Pathophysiological Conditions Rossana Roncato 1,*,†, Jacopo Angelini 2,†, Arianna Pani 2,3,†, Erika Cecchin 1, Andrea Sartore-Bianchi 2,4, Salvatore Siena 2,4, Elena De Mattia 1, Francesco Scaglione 2,3,‡ and Giuseppe Toffoli 1,‡ 1 1 Experimental and Clinical Pharmacology Unit, Centro di Riferimento Oncologico (CRO), IRCCS, 33081 Aviano, Italy; [email protected] (E.C.); [email protected] (E.D.M.); [email protected] (G.T.) 2 Department of Oncology and Hemato-Oncology, Università degli Studi di Milano, 20122 Milan, Italy; [email protected] (J.A.); [email protected] (A.P.); [email protected] (A.S-B.); [email protected] (S.S.); [email protected] (F.S.) 3 Clinical Pharmacology Unit, ASST Grande Ospedale Metropolitano Niguarda, Piazza dell'Ospedale Maggiore 3, 20162 Milan, Italy 4 Department of Hematology and Oncology, Niguarda Cancer Center, Grande Ospedale Metropolitano Niguarda, 20162 Milan, Italy * Correspondence: [email protected]; Tel.:+390434659130 † These authors contributed equally. ‡ These authors share senior authorship. Int. J. Mol. Sci. 2020, 21, x; doi: www.mdpi.com/journal/ijms Int. J. Mol. Sci. 2020, 21, x 2 of 8 Table S1. Co-administered agents categorized according to their potential risk for Drug-Drug interaction (DDI) in combination with CDK4/6 inhibitors (CDKis). Colors suggest the risk of DDI with CDKis: green, low risk DDI; orange, moderate risk DDI; red, high risk DDI. ADME, absorption, distribution, metabolism, and excretion; GI, Gastrointestinal; TdP, Torsades de Pointes; NTI, narrow therapeutic index. * Cardiological toxicity should be considered especially for ribociclib due to the QT prolongation. -

GTH 2021 State of the Art—Cardiac Surgery: the Perioperative Management of Heparin-Induced Thrombocytopenia in Cardiac Surgery
Review Article 59 GTH 2021 State of the Art—Cardiac Surgery: The Perioperative Management of Heparin-Induced Thrombocytopenia in Cardiac Surgery Laura Ranta1 Emmanuelle Scala1 1 Department of Anesthesiology, Cardiothoracic and Vascular Address for correspondence Emmanuelle Scala, MD, Centre Anesthesia, Lausanne University Hospital (CHUV), Lausanne, Hospitalier Universitaire Vaudois, Rue du Bugnon 46, BH 05/300, 1011 Switzerland Lausanne, Suisse, Switzerland (e-mail: [email protected]). Hämostaseologie 2021;41:59–62. Abstract Heparin-induced thrombocytopenia (HIT) is a severe, immune-mediated, adverse drug Keywords reaction that paradoxically induces a prothrombotic state. Particularly in the setting of ► Heparin-induced cardiac surgery, where full anticoagulation is required during cardiopulmonary bypass, thrombocytopenia the management of HIT can be highly challenging, and requires a multidisciplinary ► cardiac surgery approach. In this short review, the different perioperative strategies to run cardiopul- ► state of the art monary bypass will be summarized. Introduction genicity of the antibodies and is diagnostic for HIT. The administration of heparin to a patient with circulating Heparin-induced thrombocytopenia (HIT) is a severe, im- pathogenic HITabs puts the patient at immediate risk of mune-mediated, adverse drug reaction that paradoxically severe thrombotic complications. induces a prothrombotic state.1,2 Particularly in the setting The time course of HIT can be divided into four distinct of cardiac surgery, where full anticoagulation is required phases.6 Acute HIT is characterized by thrombocytopenia during cardiopulmonary bypass (CPB), the management of and/or thrombosis, the presence of HITabs, and confirma- HIT can be highly challenging, and requires a multidisciplin- tion of their platelet activating capacity by a functional ary approach. -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -
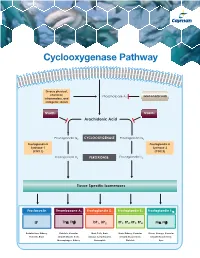
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Ventavis, INN-Iloprost
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Ventavis 10 microgram/ml nebuliser solution Ventavis 20 microgram/ml nebuliser solution 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Ventavis 10 microgram/ml nebuliser solution 1 ml solution contains 10 microgram iloprost (as iloprost trometamol). Each ampoule with 1 ml solution contains 10 microgram iloprost. Each ampoule with 2 ml solution contains 20 microgram iloprost. Ventavis 20 microgram/ml nebuliser solution 1 ml solution contains 20 microgram iloprost (as iloprost trometamol). Each ampoule with 1 ml solution contains 20 microgram iloprost. Excipient with known effect • Ventavis 10 microgram/ml: Each ml contains 0.81 mg ethanol 96% (equivalent to 0.75 mg ethanol) • Ventavis 20 microgram/ml: Each ml contains 1.62 mg ethanol 96% (equivalent to 1.50 mg ethanol). For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Nebuliser solution. Ventavis 10 microgram/ml nebuliser solution Clear, colourless solution. Ventavis 20 microgram/ml nebuliser solution Clear, colourless to slightly yellowish solution. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Treatment of adult patients with primary pulmonary hypertension, classified as NYHA functional class III, to improve exercise capacity and symptoms. 4.2 Posology and method of administration Drug product Suitable inhalation device (nebuliser) to be used Ventavis 10 microgram/ml Breelib I-Neb AAD Venta-Neb Ventavis 20 microgram/ml Breelib I-Neb AAD Ventavis should only be initiated and monitored by a physician experienced in the treatment of pulmonary hypertension. 2 Posology Dose per inhalation session At initiation of Ventavis treatment the first inhaled dose should be 2.5 microgram iloprost as delivered at the mouthpiece of the nebuliser. -

Effect of Prostanoids on Human Platelet Function: an Overview
International Journal of Molecular Sciences Review Effect of Prostanoids on Human Platelet Function: An Overview Steffen Braune, Jan-Heiner Küpper and Friedrich Jung * Institute of Biotechnology, Molecular Cell Biology, Brandenburg University of Technology, 01968 Senftenberg, Germany; steff[email protected] (S.B.); [email protected] (J.-H.K.) * Correspondence: [email protected] Received: 23 October 2020; Accepted: 23 November 2020; Published: 27 November 2020 Abstract: Prostanoids are bioactive lipid mediators and take part in many physiological and pathophysiological processes in practically every organ, tissue and cell, including the vascular, renal, gastrointestinal and reproductive systems. In this review, we focus on their influence on platelets, which are key elements in thrombosis and hemostasis. The function of platelets is influenced by mediators in the blood and the vascular wall. Activated platelets aggregate and release bioactive substances, thereby activating further neighbored platelets, which finally can lead to the formation of thrombi. Prostanoids regulate the function of blood platelets by both activating or inhibiting and so are involved in hemostasis. Each prostanoid has a unique activity profile and, thus, a specific profile of action. This article reviews the effects of the following prostanoids: prostaglandin-D2 (PGD2), prostaglandin-E1, -E2 and E3 (PGE1, PGE2, PGE3), prostaglandin F2α (PGF2α), prostacyclin (PGI2) and thromboxane-A2 (TXA2) on platelet activation and aggregation via their respective receptors. Keywords: prostacyclin; thromboxane; prostaglandin; platelets 1. Introduction Hemostasis is a complex process that requires the interplay of multiple physiological pathways. Cellular and molecular mechanisms interact to stop bleedings of injured blood vessels or to seal denuded sub-endothelium with localized clot formation (Figure1). -

Statistical Analysis Plan – Part 1
NCT03251482 Janssen Research & Development Statistical Analysis Plan – Part 1 A Randomized, Double-blind, Double-dummy, Multicenter, Adaptive Design, Dose Escalation (Part 1) and Dose-Response (Part 2) Study to Evaluate the Safety and Efficacy of Intravenous JNJ-64179375 Versus Oral Apixaban in Subjects Undergoing Elective Total Knee Replacement Surgery Protocol 64179375THR2001; Phase 2 JNJ-64179375 Status: Approved Date: 18 October 2017 Prepared by: Janssen Research & Development, LLC Document No.: EDMS-ERI-148215770 Compliance: The study described in this report was performed according to the principles of Good Clinical Practice (GCP). Confidentiality Statement The information in this document contains trade secrets and commercial information that are privileged or confidential and may not be disclosed unless such disclosure is required by applicable law or regulations. In any event, persons to whom the information is disclosed must be informed that the information is privileged or confidential and may not be further disclosed by them. These restrictions on disclosure will apply equally to all future information supplied to you that is indicated as privileged or confidential. 1 Approved, Date: 18 October 2017 JNJ-64179375 NCT03251482 Statistical Analysis Plan - Part 1 64179375THR2001 TABLE OF CONTENTS TABLE OF CONTENTS ............................................................................................................................... 2 LIST OF IN-TEXT TABLES AND FIGURES ............................................................................................... -

Antiplatelet Effects of Prostacyclin Analogues: Which One to Choose in Case of Thrombosis Or Bleeding? Sylwester P
INTERVENTIONAL CARDIOLOGY Cardiology Journal 20XX, Vol. XX, No. X, XXX–XXX DOI: 10.5603/CJ.a2020.0164 Copyright © 20XX Via Medica REVIEW ARTICLE ISSN 1897–5593 eISSN 1898–018X Antiplatelet effects of prostacyclin analogues: Which one to choose in case of thrombosis or bleeding? Sylwester P. Rogula1*, Hubert M. Mutwil1*, Aleksandra Gąsecka1, Marcin Kurzyna2, Krzysztof J. Filipiak1 11st Chair and Department of Cardiology, Medical University of Warsaw, Poland 2Department of Pulmonary Circulation, Thromboembolic Diseases and Cardiology, Center of Postgraduate Education Medical, European Health Center Otwock, Poland Abstract Prostacyclin and analogues are successfully used in the treatment of pulmonary arterial hypertension (PAH) due to their vasodilatory effect on pulmonary arteries. Besides vasodilatory effect, prostacyclin analogues inhibit platelets, but their antiplatelet effect is not thoroughly established. The antiplatelet effect of prostacyclin analogues may be beneficial in case of increased risk of thromboembolic events, or undesirable in case of increased risk of bleeding. Since prostacyclin and analogues differ regarding their potency and form of administration, they might also inhibit platelets to a different extent. This review summarizes the recent evidence on the antiplatelet effects of prostacyclin and analogue in the treatment of PAH, this is important to consider when choosing the optimal treatment regimen in tailoring to an individual patients’ needs. (Cardiol J 20XX; XX, X: xx–xx) Key words: prostacyclin analogues, pulmonary arterial hypertension, platelets, antiplatelet effect, thrombosis, bleeding Introduction tiproliferative effects [4]. The main indication for PGI2 and analogues is advanced pulmonary arterial Since 1935 when prostaglandin was isolated hypertension (PAH) and peripheral vascular disor- for the first time [1], many scientists have focused ders [5]. -

2374 Supplementary Drugs and Other Substances
2374 Supplementary Drugs and Other Substances 5. Burdock GA. Review of the biological properties and toxicity of Pharmacokinetics reduced to the endoperoxide prostaglandin H2 (PGH2). Prostag- bee propolis (propolis). Food Chem Toxicol 1998; 36: 347–63. Propylene glycol is rapidly absorbed from the gastrointestinal landin H2 is then converted to the primary prostaglandins pros- 6. Lieberman HD, et al. Allergic contact dermatitis to propolis in a tract. There is evidence of topical absorption when applied to taglandin D2, prostaglandin E2, and prostaglandin F2 , to throm- violin maker. J Am Acad Dermatol 2002; 46 (suppl): S30–S31. damaged skin. boxane A2 (TXA2) via the enzyme thromboxane synthetase, or 7. Giusti F, et al. Sensitization to propolis in 1255 children under- It is extensively metabolised in the liver primarily by oxidation to prostacyclin (PGI2) via the enzyme prostacyclin synthetase. going patch testing. Contact Dermatitis 2004; 51: 255–8. to lactic and pyruvic acid and is also excreted in the urine These products are further metabolised and rapidly inactivated in 8. Walgrave SE, et al. Allergic contact dermatitis from propolis. unchanged. the body. Dermatitis 2005; 16: 209–15. The secondary prostaglandins, prostaglandin A (PGA ), pros- 9. Majiene D, et al. Antifungal and antibacterial activity of propo- ◊ References. 2 2 lis. Curr Nutr Food Sci 2007; 3: 304–8. 1. Yu DK, et al. Pharmacokinetics of propylene glycol in humans taglandin B2 (PGB2), and prostaglandin C2 (PGC2) are derived during multiple dosing regimens. J Pharm Sci 1985; 74: 876–9. from prostaglandin E2, but are formed during extraction and Preparations 2. Speth PAJ, et al.