Profiling Microdissected Epithelium and Stroma to Model Genomic Signatures for Cervical Carcinogenesis Accommodating for Covariates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Key Genomic Signature Associated with Lymphovascular Invasion in Head and Neck Squamous Cell Carcinoma
A key genomic signature associated with lymphovascular invasion in head and neck squamous cell carcinoma Jian Zhang Aliated Cancer hospital & Institute of Guangzhou Medical University Huali Jiang Aliated Donghua Hospital of Sun Yat-sen University Tao Xie Aliated Cancer Hospital of Guangzhou Medical University Baiyao Wang Aliated Cancer Hospital of Guangzhou Medical Unversity Xiaoting Huang Aliated Cancer Hospital & Institute of Guangzhou Medical University Jie Lin Aliated Cancer Hospital & Institute of Guangzhou Medical University Anan Xu Aliated Cancer Hospital of Guangzhou Medical University Rong Li Aliated Cancer Hospital & Institute of Guangzhou Medical University Yawei Yuan ( [email protected] ) Guangzhou Medical University Aliated Cancer Hospital Research article Keywords: lymphovascular invasion, head and neck squamous cell carcinoma, hub genes, TCGA, weighted gene co-expression network analysis Posted Date: January 16th, 2020 DOI: https://doi.org/10.21203/rs.2.18349/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/24 Abstract Objective: Lymphovascular invasion (LOI), a key pathological feature of head and neck squamous cell carcinoma (HNSCC), predicts poor survival. However, the associated clinical characteristics remain uncertain, and the molecular mechanisms are largely unknown. Methods: Weighted gene co-expression network analysis was performed to construct gene co-expression networks and investigate the relationship between modules and LOI clinical trait. Functional enrichment and KEGG pathway enrichment analysis were performed for differentially expressed genes using DAVID database. The protein-protein interaction network was constructed using Cytoscape software, and module analysis was performed using MCODE. Prognosis role and expression analysis was further validated by survival analysis, GEPIA analysis and HPA database. -

Reactive Stroma in Human Prostate Cancer: Induction of Myofibroblast Phenotype and Extracellular Matrix Remodeling1
2912 Vol. 8, 2912–2923, September 2002 Clinical Cancer Research Reactive Stroma in Human Prostate Cancer: Induction of Myofibroblast Phenotype and Extracellular Matrix Remodeling1 Jennifer A. Tuxhorn, Gustavo E. Ayala, Conclusions: The stromal microenvironment in human Megan J. Smith, Vincent C. Smith, prostate cancer is altered compared with normal stroma and Truong D. Dang, and David R. Rowley2 exhibits features of a wound repair stroma. Reactive stroma is composed of myofibroblasts and fibroblasts stimulated to Departments of Molecular and Cellular Biology [J. A. T., T. D. D., express extracellular matrix components. Reactive stroma D. R. R.] and Pathology [G. E. A., M. J. S., V. C. S.] Baylor College of Medicine, Houston, Texas 77030 appears to be initiated during PIN and evolve with cancer progression to effectively displace the normal fibromuscular stroma. These studies and others suggest that TGF-1isa ABSTRACT candidate regulator of reactive stroma during prostate can- Purpose: Generation of a reactive stroma environment cer progression. occurs in many human cancers and is likely to promote tumorigenesis. However, reactive stroma in human prostate INTRODUCTION cancer has not been defined. We examined stromal cell Activation of the host stromal microenvironment is pre- phenotype and expression of extracellular matrix compo- dicted to be a critical step in adenocarcinoma growth and nents in an effort to define the reactive stroma environ- progression (1–5). Several human cancers have been shown to ment and to determine its ontogeny during prostate cancer induce a stromal reaction or desmoplasia as a component of progression. carcinoma progression. However, the specific mechanisms of Experimental Design: Normal prostate, prostatic intra- stromal cell activation are not known, and the extent to which epithelial neoplasia (PIN), and prostate cancer were exam- stroma regulates the biology of tumorigenesis is not fully un- ined by immunohistochemistry. -

Apoptotic Genes As Potential Markers of Metastatic Phenotype in Human Osteosarcoma Cell Lines
17-31 10/12/07 14:53 Page 17 INTERNATIONAL JOURNAL OF ONCOLOGY 32: 17-31, 2008 17 Apoptotic genes as potential markers of metastatic phenotype in human osteosarcoma cell lines CINZIA ZUCCHINI1, ANNA ROCCHI2, MARIA CRISTINA MANARA2, PAOLA DE SANCTIS1, CRISTINA CAPANNI3, MICHELE BIANCHINI1, PAOLO CARINCI1, KATIA SCOTLANDI2 and LUISA VALVASSORI1 1Dipartimento di Istologia, Embriologia e Biologia Applicata, Università di Bologna, Via Belmeloro 8, 40126 Bologna; 2Laboratorio di Ricerca Oncologica, Istituti Ortopedici Rizzoli; 3IGM-CNR, Unit of Bologna, c/o Istituti Ortopedici Rizzoli, Via di Barbiano 1/10, 40136 Bologna, Italy Received May 29, 2007; Accepted July 19, 2007 Abstract. Metastasis is the most frequent cause of death among malignant primitive bone tumor, usually developing in children patients with osteosarcoma. We have previously demonstrated and adolescents, with a high tendency to metastasize (2). in independent experiments that the forced expression of Metastases in osteosarcoma patients spread through peripheral L/B/K ALP and CD99 in U-2 OS osteosarcoma cell lines blood very early and colonize primarily the lung, and later markedly reduces the metastatic ability of these cancer cells. other skeleton districts (3). Since disseminated hidden micro- This behavior makes these cell lines a useful model to assess metastases are present in 80-90% of OS patients at the time the intersection of multiple and independent gene expression of diagnosis, the identification of markers of invasiveness signatures concerning the biological problem of dissemination. and metastasis forms a target of paramount importance in With the aim to characterize a common transcriptional profile planning the treatment of osteosarcoma lesions and enhancing reflecting the essential features of metastatic behavior, we the prognosis. -

Colposcopy of the Uterine Cervix
THE CERVIX: Colposcopy of the Uterine Cervix • I. Introduction • V. Invasive Cancer of the Cervix • II. Anatomy of the Uterine Cervix • VI. Colposcopy • III. Histology of the Normal Cervix • VII: Cervical Cancer Screening and Colposcopy During Pregnancy • IV. Premalignant Lesions of the Cervix The material that follows was developed by the 2002-04 ASCCP Section on the Cervix for use by physicians and healthcare providers. Special thanks to Section members: Edward J. Mayeaux, Jr, MD, Co-Chair Claudia Werner, MD, Co-Chair Raheela Ashfaq, MD Deborah Bartholomew, MD Lisa Flowers, MD Francisco Garcia, MD, MPH Luis Padilla, MD Diane Solomon, MD Dennis O'Connor, MD Please use this material freely. This material is an educational resource and as such does not define a standard of care, nor is intended to dictate an exclusive course of treatment or procedure to be followed. It presents methods and techniques of clinical practice that are acceptable and used by recognized authorities, for consideration by licensed physicians and healthcare providers to incorporate into their practice. Variations of practice, taking into account the needs of the individual patient, resources, and limitation unique to the institution or type of practice, may be appropriate. I. AN INTRODUCTION TO THE NORMAL CERVIX, NEOPLASIA, AND COLPOSCOPY The uterine cervix presents a unique opportunity to clinicians in that it is physically and visually accessible for evaluation. It demonstrates a well-described spectrum of histological and colposcopic findings from health to premalignancy to invasive cancer. Since nearly all cervical neoplasia occurs in the presence of human papillomavirus infection, the cervix provides the best-defined model of virus-mediated carcinogenesis in humans to date. -

A Dissertation Entitled the Androgen Receptor
A Dissertation entitled The Androgen Receptor as a Transcriptional Co-activator: Implications in the Growth and Progression of Prostate Cancer By Mesfin Gonit Submitted to the Graduate Faculty as partial fulfillment of the requirements for the PhD Degree in Biomedical science Dr. Manohar Ratnam, Committee Chair Dr. Lirim Shemshedini, Committee Member Dr. Robert Trumbly, Committee Member Dr. Edwin Sanchez, Committee Member Dr. Beata Lecka -Czernik, Committee Member Dr. Patricia R. Komuniecki, Dean College of Graduate Studies The University of Toledo August 2011 Copyright 2011, Mesfin Gonit This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract of The Androgen Receptor as a Transcriptional Co-activator: Implications in the Growth and Progression of Prostate Cancer By Mesfin Gonit As partial fulfillment of the requirements for the PhD Degree in Biomedical science The University of Toledo August 2011 Prostate cancer depends on the androgen receptor (AR) for growth and survival even in the absence of androgen. In the classical models of gene activation by AR, ligand activated AR signals through binding to the androgen response elements (AREs) in the target gene promoter/enhancer. In the present study the role of AREs in the androgen- independent transcriptional signaling was investigated using LP50 cells, derived from parental LNCaP cells through extended passage in vitro. LP50 cells reflected the signature gene overexpression profile of advanced clinical prostate tumors. The growth of LP50 cells was profoundly dependent on nuclear localized AR but was independent of androgen. Nevertheless, in these cells AR was unable to bind to AREs in the absence of androgen. -

Plasma Cells in Vitro Generation of Long-Lived Human
Downloaded from http://www.jimmunol.org/ by guest on September 24, 2021 is online at: average * The Journal of Immunology , 32 of which you can access for free at: 2012; 189:5773-5785; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication November 2012; doi: 10.4049/jimmunol.1103720 http://www.jimmunol.org/content/189/12/5773 In Vitro Generation of Long-lived Human Plasma Cells Mario Cocco, Sophie Stephenson, Matthew A. Care, Darren Newton, Nicholas A. Barnes, Adam Davison, Andy Rawstron, David R. Westhead, Gina M. Doody and Reuben M. Tooze J Immunol cites 65 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2012/11/16/jimmunol.110372 0.DC1 This article http://www.jimmunol.org/content/189/12/5773.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 24, 2021. The Journal of Immunology In Vitro Generation of Long-lived Human Plasma Cells Mario Cocco,*,1 Sophie Stephenson,*,1 Matthew A. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

KIF23 Enhances Cell Proliferation in Pancreatic Ductal Adenocarcinoma and Is a Potent Therapeutic Target
1394 Original Article Page 1 of 15 KIF23 enhances cell proliferation in pancreatic ductal adenocarcinoma and is a potent therapeutic target Chun-Tao Gao1#, Jin Ren2#, Jie Yu1,3#, Sheng-Nan Li1, Xiao-Fan Guo1, Yi-Zhang Zhou1 1Department of Pancreatic Cancer, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Tianjin Key Laboratory of Cancer Prevention and Therapy, Tianjin’s Clinical Research Center for Cancer, Tianjin, China; 2Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Taiyuan, China; 3The First Hospital of Shanxi Medical University, Taiyuan, China Contributions: (I) Conception and design: CT Gao, J Ren; (II) Administrative support: CT Gao; (III) Provision of study materials or patients: J Yu; (IV) Collection and assembly of data: J Ren, SN Li, YZ Zhou; (V) Data analysis and interpretation: XF Guo, J Ren; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Chun-Tao Gao. Department of Pancreatic Cancer, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Tianjin Key Laboratory of Cancer Prevention and Therapy, Tianjin’s Clinical Research Center for Cancer, Huan-hu-xi Road, He-xi District, Tianjin 300060, China. Email: [email protected]. Background: In recent research, high expression of kinesin family member 23 (KIF23), one of the kinesin motor proteins involved in the regulation of cytokinesis, has been shown to be related to poor prognosis in glioma and paclitaxel-resistant gastric cancer, as a results of the enhancement of proliferation, migration, and invasion. In this study, we analyzed the role of KIF23 in the progression of pancreatic ductal adenocarcinoma. -

Tumor-Associated Stromal Cells As Key Contributors to the Tumor Microenvironment Karen M
Bussard et al. Breast Cancer Research (2016) 18:84 DOI 10.1186/s13058-016-0740-2 REVIEW Open Access Tumor-associated stromal cells as key contributors to the tumor microenvironment Karen M. Bussard1,2, Lysette Mutkus3, Kristina Stumpf3, Candelaria Gomez-Manzano4 and Frank C. Marini1,3* Abstract The tumor microenvironment is a heterogeneous population of cells consisting of the tumor bulk plus supporting cells. It is becoming increasingly evident that these supporting cells are recruited by cancer cells from nearby endogenous host stroma and promote events such as tumor angiogenesis, proliferation, invasion, and metastasis, as well as mediate mechanisms of therapeutic resistance. In addition, recruited stromal cells range in type and include vascular endothelial cells, pericytes, adipocytes, fibroblasts, and bone-marrow mesenchymal stromal cells. During normal wound healing and inflammatory processes, local stromal cells change their phenotype to become that of reactive stroma. Under certain conditions, however, tumor cells can co-opt these reactive stromal cells and further transition them into tumor-associated stromal cells (TASCs). These TASCs express higher levels of proteins, including alpha-smooth muscle actin, fibroblast activating protein, and matrix metalloproteinases, compared with their normal, non-reactive counterparts. TASCs are also known to secrete many pro-tumorigenic factors, including IL-6, IL-8, stromal-derived factor-1 alpha, vascular endothelial growth factor, tenascin-C, and matrix metalloproteinases, among others, which recruit additional tumor and pro-tumorigenic cells to the developing microenvironment. Here, we review the current literature pertaining to the origins of recruited host stroma, contributions toward tumor progression, tumor-associated stromal cells, and mechanisms of crosstalk between endogenous host stroma and tumor cells. -
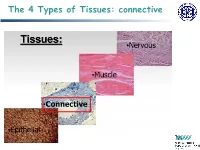
The 4 Types of Tissues: Connective
The 4 Types of Tissues: connective Connective Tissue General structure of CT cells are dispersed in a matrix matrix = a large amount of extracellular material produced by the CT cells and plays a major role in the functioning matrix component = ground substance often crisscrossed by protein fibers ground substance usually fluid, but it can also be mineralized and solid (bones) CTs = vast variety of forms, but typically 3 characteristic components: cells, large amounts of amorphous ground substance, and protein fibers. Connective Tissue GROUND SUBSTANCE In connective tissue, the ground substance is an amorphous gel-like substance surrounding the cells. In a tissue, cells are surrounded and supported by an extracellular matrix. Ground substance traditionally does not include fibers (collagen and elastic fibers), but does include all the other components of the extracellular matrix . The components of the ground substance vary depending on the tissue. Ground substance is primarily composed of water, glycosaminoglycans (most notably hyaluronan ), proteoglycans, and glycoproteins. Usually it is not visible on slides, because it is lost during the preparation process. Connective Tissue Functions of Connective Tissues Support and connect other tissues Protection (fibrous capsules and bones that protect delicate organs and, of course, the skeletal system). Transport of fluid, nutrients, waste, and chemical messengers is ensured by specialized fluid connective tissues, such as blood and lymph. Adipose cells store surplus energy in the form of fat and contribute to the thermal insulation of the body. Embryonic Connective Tissue All connective tissues derive from the mesodermal layer of the embryo . The first connective tissue to develop in the embryo is mesenchyme , the stem cell line from which all connective tissues are later derived. -

How Does SUMO Participate in Spindle Organization?
cells Review How Does SUMO Participate in Spindle Organization? Ariane Abrieu * and Dimitris Liakopoulos * CRBM, CNRS UMR5237, Université de Montpellier, 1919 route de Mende, 34090 Montpellier, France * Correspondence: [email protected] (A.A.); [email protected] (D.L.) Received: 5 July 2019; Accepted: 30 July 2019; Published: 31 July 2019 Abstract: The ubiquitin-like protein SUMO is a regulator involved in most cellular mechanisms. Recent studies have discovered new modes of function for this protein. Of particular interest is the ability of SUMO to organize proteins in larger assemblies, as well as the role of SUMO-dependent ubiquitylation in their disassembly. These mechanisms have been largely described in the context of DNA repair, transcriptional regulation, or signaling, while much less is known on how SUMO facilitates organization of microtubule-dependent processes during mitosis. Remarkably however, SUMO has been known for a long time to modify kinetochore proteins, while more recently, extensive proteomic screens have identified a large number of microtubule- and spindle-associated proteins that are SUMOylated. The aim of this review is to focus on the possible role of SUMOylation in organization of the spindle and kinetochore complexes. We summarize mitotic and microtubule/spindle-associated proteins that have been identified as SUMO conjugates and present examples regarding their regulation by SUMO. Moreover, we discuss the possible contribution of SUMOylation in organization of larger protein assemblies on the spindle, as well as the role of SUMO-targeted ubiquitylation in control of kinetochore assembly and function. Finally, we propose future directions regarding the study of SUMOylation in regulation of spindle organization and examine the potential of SUMO and SUMO-mediated degradation as target for antimitotic-based therapies. -

Genomics and Functional Genomics of Malignant Pleural Mesothelioma
International Journal of Molecular Sciences Review Genomics and Functional Genomics of Malignant Pleural Mesothelioma Ece Cakiroglu 1,2 and Serif Senturk 1,2,* 1 Izmir Biomedicine and Genome Center, Izmir 35340, Turkey; [email protected] 2 Department of Genome Sciences and Molecular Biotechnology, Izmir International Biomedicine and Genome Institute, Dokuz Eylul University, Izmir 35340, Turkey * Correspondence: [email protected] Received: 22 July 2020; Accepted: 20 August 2020; Published: 1 September 2020 Abstract: Malignant pleural mesothelioma (MPM) is a rare, aggressive cancer of the mesothelial cells lining the pleural surface of the chest wall and lung. The etiology of MPM is strongly associated with prior exposure to asbestos fibers, and the median survival rate of the diagnosed patients is approximately one year. Despite the latest advancements in surgical techniques and systemic therapies, currently available treatment modalities of MPM fail to provide long-term survival. The increasing incidence of MPM highlights the need for finding effective treatments. Targeted therapies offer personalized treatments in many cancers. However, targeted therapy in MPM is not recommended by clinical guidelines mainly because of poor target definition. A better understanding of the molecular and cellular mechanisms and the predictors of poor clinical outcomes of MPM is required to identify novel targets and develop precise and effective treatments. Recent advances in the genomics and functional genomics fields have provided groundbreaking insights into the genomic and molecular profiles of MPM and enabled the functional characterization of the genetic alterations. This review provides a comprehensive overview of the relevant literature and highlights the potential of state-of-the-art genomics and functional genomics research to facilitate the development of novel diagnostics and therapeutic modalities in MPM.