Catalytic Approaches to the Synthesis of Amide Bonds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lecture 10 Carbon-Nitrogen Bonds Formation I
NPTEL – Chemistry – Principles of Organic Synthesis Lecture 10 Carbon-Nitrogen Bonds Formation I 4.1 Principles The methods for the formation of bonds between nitrogen and aliphatic carbon can be broadly divided into two categories: (i) reaction of nucleophilic nitrogen with electrophilic carbon, and (ii) reaction of electrophilic nitrogen with nucleophilic carbon. In this section, we will try to cover some of the important reactions. 4.2 Substitution of Nitrogen Nucleophiles at Saturated Carbon 4.2.1 Ritter Reaction Treatment of a tertiary alcohol or the corresponding alkene with concentrated sulphuric acid and a nitrile affords an amide that in acidic conditions undergoes hydrolysis to give amine. First, a R H, RCN H3O + RCO H OH N O NH 2 H2O H 2 Mechanism OH2 H -H2O :NC-R H2O OH OH N N R N R 2 C R OH O H2O proton H3O + RCO2H N R N R NH2 -H3O transfer H Scheme 1 tertiary carbocation is formed which is attacked by the nitrile to give a quaternary ion. The latter is decomposed by water to provide an amide which, in acidic conditions, is hydrolyzed to give an amine (Scheme 1). Joint initiative of IITs and IISc – Funded by MHRD Page 1 of 35 NPTEL – Chemistry – Principles of Organic Synthesis Examples: Me H N H2SO4 + N Chlorobenzene O 91% S. J. Chang, Organic Process Research and Development 1999, 3, 232. Me Me Me Me Me Me Me Me SnCl4 MeCN CH2 CH2 Cl N N T.-L. Ho, R.-J. Chein, J. Org. Chem. 2004, 69, 591. Me Me O Ph HNCHO Ph OH Ph HN H2SO4 Me H2SO4 N TMSCN N MeCN N Bn Bn Bn H. -

Nomenclature of Carboxylic Acids • Select the Longest Carbon Chain Containing the Carboxyl Group
Chapter 5 Carboxylic Acids and Esters Carboxylic Acids • Carboxylic acids are weak organic acids which Chapter 5 contain the carboxyl group (RCO2H): Carboxylic Acids and Esters O C O H O RCOOH RCO2H Chapter Objectives: O condensed ways of • Learn to recognize the carboxylic acid, ester, and related functional groups. RCOH writing the carboxyl • Learn the IUPAC system for naming carboxylic acids and esters. group a carboxylic acid C H • Learn the important physical properties of the carboxylic acids and esters. • Learn the major chemical reaction of carboxylic acids and esters, and learn how to O predict the products of ester synthesis and hydrolysis reactions. the carboxyl group • Learn some of the important properties of condensation polymers, especially the polyesters. Mr. Kevin A. Boudreaux • The tart flavor of sour-tasting foods is often caused Angelo State University CHEM 2353 Fundamentals of Organic Chemistry by the presence of carboxylic acids. Organic and Biochemistry for Today (Seager & Slabaugh) www.angelo.edu/faculty/kboudrea 2 Nomenclature of Carboxylic Acids • Select the longest carbon chain containing the carboxyl group. The -e ending of the parent alkane name is replaced by the suffix -oic acid. • The carboxyl carbon is always numbered “1” but the number is not included in the name. • Name the substituents attached to the chain in the Nomenclature of usual way. • Aromatic carboxylic acids (i.e., with a CO2H Carboxylic Acids directly connected to a benzene ring) are named after the parent compound, benzoic acid. O C OH 3 -

Optimisation Du Métabolisme Énergétique Du Soufre Chez La Bactérie Hyperthermophile Aquifex Aeolicus
Aix-Marseille Université Ecole Doctorale des Sciences de la Vie et de la Santé Thèse de Doctorat d’Université Mention Microbiologie Clément AUSSIGNARGUES Optimisation du métabolisme énergétique du soufre chez la bactérie hyperthermophile Aquifex aeolicus Soutenue le 17 Décembre 2012 devant le jury: Pr. Frédéric BARRAS Dr. Laurent COURNAC Dr. Bruno FRANZETTI Dr. Marie-Thérèse GIUDICI-ORTICONI Dr. Anne GODFROY Dr. Marianne ILBERT Laboratoire de Bioénergétique et Ingénierie des Protéines Centre National de la Recherche Scientifique Aix-Marseille Université Ecole Doctorale des Sciences de la Vie et de la Santé Thèse de Doctorat d’Université Mention Microbiologie Clément AUSSIGNARGUES Optimisation du métabolisme énergétique du soufre chez la bactérie hyperthermophile Aquifex aeolicus Soutenue le 17 Décembre 2012 devant le jury: Pr. Frédéric BARRAS Dr. Laurent COURNAC Dr. Bruno FRANZETTI Dr. Marie-Thérèse GIUDICI-ORTICONI Dr. Anne GODFROY Dr. Marianne ILBERT Laboratoire de Bioénergétique et Ingénierie des Protéines Centre National de la Recherche Scientifique Sommaire Sommaire Introduction 2 Chapitre I- Métabolismes énergétiques du soufre 4 I) Le soufre, un élément essentiel aux multiples facettes 6 II) Le soufre utilisé comme composé énergétique 9 A) Oxydation des composés du soufre 10 1. Oxydation du sulfure d’hydrogène H2S 10 a) La Sulfure Quinone Oxydoréductase (SQR) 10 b) La flavocytochrome c sulfure déshydrogénase (FCSD) 13 2- 2. Oxydation du thiosulfate (S2O3 ) 14 a) Thiosulfate : accepteur oxydoréductase (TAOR) 14 b) Le système Sox 16 2- 3. Oxydation du sulfite (SO3 ) 20 a) Sulfite : accepteur oxydoréductase (SAOR) 21 b) Voie de l’APS 23 4. Oxydation du soufre stocké dans les globules 26 B) La réduction du soufre : soufre/polysulfure réductase 29 C) Un cas particulier : la dismutation du soufre (SOR) 33 1. -

MDA) Analogs and Homologs
Terry A. Dal Cason, 1 M.Sc. An Evaluation of the Potential for Clandestine Manufacture of 3,4-Methylenedioxyamphetamine (MDA) Analogs and Homologs REFERENCE: Dal Cason, T. A., "An Evaluation of the Potential for Clandestine Manu- facture of 3,4-Methylenedioxyamphetamine (MDA) Analogs and Homologs," Journal of Forensic Sciences, JFSCA, Vol. 35, No.3, May 1990, pp. 675-697. ABSTRACT: Encountering a novel controlled-substance analog (designer drug) has become a distinct possibility for all forensic drug laboratories. 3,4-Methylenedioxyamphetamine (MDA) in particular is a receptive parent compound for the molecular modifications which produce such homo logs and analogs. The identification of these compounds, however, can prove to be an arduous task. It would be desirable to direct the focus of the identification to those compounds which are the more likely candidates for clandestine-laboratory synthesis. The process of narrowing the range of theoretical possibilities to logical choices may be enhanced by using a suitable predictive scheme. Such a predictive scheme for MDA analogs is presented based on putative Central Nervous System activity, existence or formulation of a reasonable synthesis method, and availability of the required precursors. KEYWORDS: toxicology, 3,4-methylenedioxyamphetamine, drug identification To circumvent statutes enacted to control the use of various dangerous drugs (controlled substances), clandestine laboratory operators will sometimes make minor alterations in the molecular structure of a parent compound. These structural changes are reflected in the chemical nomenclature of the new analog or homolog, and, in the past/ had effectively removed it from the purview of the law. Such modifications, at least in the case of 3,4- methylenedioxyamphetamine (MDA), do not appear to be haphazard. -

And Intermolecular Addition of Carboxylic Acids to Alkynes in Aqueous Media: a Review
catalysts Review Metal-Catalyzed Intra- and Intermolecular Addition of Carboxylic Acids to Alkynes in Aqueous Media: A Review Javier Francos and Victorio Cadierno * Laboratorio de Compuestos Organometálicos y Catálisis (Unidad Asociada al CSIC), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Departamento de Química Orgánica e Inorgánica, IUQOEM, Facultad de Química, Universidad de Oviedo, Julián Clavería 8, E-33006 Oviedo, Spain; [email protected] * Correspondence: [email protected]; Tel.: +34-985-103453 Received: 19 October 2017; Accepted: 2 November 2017; Published: 6 November 2017 Abstract: The metal-catalyzed addition of carboxylic acids to alkynes is a very effective tool for the synthesis of carboxylate-functionalized olefinic compounds in an atom-economical manner. Thus, a large variety of synthetically useful lactones and enol-esters can be accessed through the intra- or intermolecular versions of this process. In order to reduce the environmental impact of these reactions, considerable efforts have been devoted in recent years to the development of catalytic systems able to operate in aqueous media, which represent a real challenge taking into account the tendency of alkynes to undergo hydration in the presence of transition metals. Despite this, different Pd, Pt, Au, Cu and Ru catalysts capable of promoting the intra- and intermolecular addition of carboxylic acids to alkynes in a selective manner in aqueous environments have appeared in the literature. In this review article, an overview of this chemistry is provided. The synthesis of b-oxo esters by catalytic addition of carboxylic acids to terminal propargylic alcohols in water is also discussed. Keywords: aqueous catalysis; alkynes; carboxylic acids; alkynoic acids; hydrocarboxylation reactions; cycloisomerization reactions; enol esters; lactones; b-oxo esters 1. -

Carboxylic Acid Addition to Terminal Alkynes Utilizing Ammonium Tagged
Journal of Organometallic Chemistry 883 (2019) 11e16 Contents lists available at ScienceDirect Journal of Organometallic Chemistry journal homepage: www.elsevier.com/locate/jorganchem Carboxylic acid addition to terminal alkynes utilizing ammonium tagged Hoveyda-Grubbs catalyst supported on magnetically separable core/shell silica: A highly reusable and air compatible catalytic system € € * ** Bengi Ozgün Oztürk , Didar Gürcü, Solmaz Karabulut S¸ ehitoglu Hacettepe University, Faculty of Science, Chemistry Department, 06800, Beytepe, Ankara, Turkey article info abstract Article history: In this study, the performance of ammonium tagged Hoveyda-Grubbs catalyst supported on magnetically Received 24 October 2018 separable core/shell silica gel was tested on carboxylic acid addition reactions to terminal alkynes using a Received in revised form variety of carboxylic acid derivatives under air atmosphere. The catalytic system was found to be 13 December 2018 compatible with air atmosphere and can tolerate even non-degassed solvents. The reaction parameters Accepted 10 January 2019 such as temperature, substrate/catalyst ratio and the effect of carboxylic acid on the selectivity and yield Available online 14 January 2019 of the reaction were investigated in details. The reaction of arylacetylenes with acetic acid yielded the corresponding E-isomer with conversion values up to 99% with a catalytic loading of 1% Ru. The reus- Keywords: Carboxylic acid ability of the catalyst was tested using acetic acid/benzoic acid and phenylacetylene in toluene at 85 C Alkyne under air atmosphere. The catalyst was found to be highly reusable and maintained its activity up to 11th Ruthenium run, reaching a conversion value of 83% with minimum ruthenium leaching. Reusable catalyst © 2019 Elsevier B.V. -

Catalytic Radical-Polar Crossover Ritter Reaction Eric E
Catalytic Radical-Polar Crossover Ritter Reaction Eric E. Touney‡, Riley E. Cooper‡, Sarah. E. Bredenkamp, David T. George†, and Sergey V. Pronin* Department of Chemistry, University of California, Irvine, California 92697-2025, United States Supporting Information Placeholder Co(II) complex, ABSTRACT: A catalytic radical-polar crossover Ritter R3 R3 R3 silane, oxidant MeCN, H2O R1 R1 AcHN reaction is described. The transformation proceeds under R4 R4 R4 via R3 nuclephilic attack 1 2 acid-free conditions and tolerates a variety of functional R2 R2 & hydrolysis R R R1 R4 groups. The catalyst design overcomes limitations in the 32 examples substitution pattern of starting materials and enables hy- R2 droamidation of a diverse range of alkenes. Formation of Scheme 1. Catalytic radical-polar crossover Ritter reaction. hydrogen contributes to the background consumption of reductant and oxidant and competes with the desired path- polar crossover reactions of alkenes we observed that ap- way, pointing to a mechanistic link between hydrogen plication of acetonitrile as solvent occasionally resulted atom transfer-initiated organic reactions and hydrogen in formation of small amounts of side-products arising evolution catalysis. from apparent Markovnikov addition of acetamide to the carbon-carbon double bond.5,6 The electrophilic interme- diates in this formal Ritter reaction were proposed to arise from direct oxidation of alkyl radicals following the HAT Hydroamidation of alkenes with nitriles in the presence and diffusion from the solvent cage.7 Alternative path- of a strong Brønsted acid, discovered by Ritter, allows for ways involve radical pair collapse to generate the corre- a convenient access to tert-alkylamines and their deriva- 1 sponding alkylcobalt(III) complexes, which undergo oxi- tives from simple precursors. -
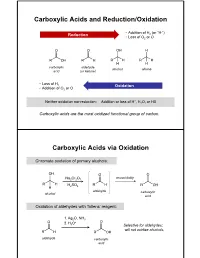
Carboxylic Acids and Reduction/Oxidation Carboxylic
Carboxylic Acids and Reduction/Oxidation • Addition of H (or “H-”) Reduction 2 • Loss of O2 or O carboxylic aldehyde alcohol alkane acid (or ketone) • Loss of H 2 Oxidation • Addition of O2 or O + Neither oxidation nor reduction: Addition or loss of H , H2O, or HX Carboxylic acids are the most oxidized functional group of carbon. Carboxylic Acids via Oxidation Chromate oxidation of primary alcohols: unavoidably Na2Cr2O7 H2SO4 aldehyde carboxylic alcohol acid Oxidation of aldehydes with Tollens’ reagent: 1. Ag2O, NH3 2. H O+ 3 Selective for aldehydes; will not oxidize alcohols. aldehyde carboxylic acid Carboxylic Acids via Oxidation Oxidative cleavage of alkynes: 1. O3 2. H2O internal alkyne Oxidation of benzylic carbons: KMnO4 Purifying Carboxylic Acids by Acid-Base Extraction Acidic and basic organic molecules can be separated from other substances by manipulating their protonation state and solubility. Let’s say we run a reaction. Na2Cr2O7 + unreacted H2SO4 + Na+ 3+ We want this… + Cr(H2O)6 + organic + HSO - …but not these. side products 4 How do we isolate our desired product from the other materials, without using distillation or chromatography? Acid-Base Extraction Step 1: Remove inorganics from organics via differential solubility. a separatory (sept) funnel - Na+ HSO4 Cr(H O) 3+ H2O 2 6 CHCl3 + organic side densities: products (H2O) = 1 g/mL (CHCl3) = 1.48 g/mL A large density difference ensures the two liquids separate completely and quickly. (Much faster than, say, oil and water.) Step 2: Add basic water to deprotonate carboxylic acid, and transfer it to the aqueous phase. - Na+ HSO4 3+ discard Cr(H2O)6 H2O layer + organic + organic side side products products add discard NaOH/ H2O CHCl3 layer + organic side products Step 3: Re-acidify carboxylate to return it to organic solution. -

Chapter 20: Carboxylic Acids and Nitriles
CHAPTER 20 CARBOXYLIC ACIDS Based on McMurry’s Organic Chemistry, 6th edition Dr.Mohanad Al-Hachamii The Importance of Carboxylic Acids (RCO2H) Starting materials for acyl derivatives (esters, amides, and acid chlorides) Abundant in nature from oxidation of aldehydes and alcohols in metabolism Acetic acid, CH3CO2H, - vinegar Butanoic acid, CH3CH2CH2CO2H (rancid butter) Long-chain aliphatic acids from the breakdown of fats Based on McMurry, Organic Chemistry, Chapter 20, 6th edition, (c) 2003 Dr.Mohanad Al-Hachamii 2 20.1 Naming Carboxylic Acids •Carboxylic Acids, RCO2H •If derived from open-chain alkanes, replace the terminal -e of the alkane name with -oic acid •The carboxyl carbon atom is C1 Based on McMurry, Organic Chemistry, Chapter 20, 6th Dr.Mohanad Al-Hachamii edition, (c) 2003 3 Alternative Names Compounds with CO2H bonded to a ring are named using the suffix -carboxylic acid The CO2H carbon is not itself numbered in this system Use common names for formic acid (HCOOH) and acetic acid (CH3COOH) – see Table 20.1 Based on McMurry, Organic Chemistry, Chapter 20, 6th edition, (c) 2003 Dr.Mohanad Al-Hachamii 4 5 • Greek letters are used to designate the location of substituents in common names. • The carbon adjacent to the COOH is called the carbon, followed by the carbon, followed by the carbon, the carbon and so forth down the chain. • The last carbon in the chain is sometimes called the carbon. • The carbon in the common system is numbered C2 in the IUPAC system. 6 • Compounds containing two carboxy groups are called diacids. Diacids are named using the suffix –dioic acid. -

Amidation of Carboxylic Acids with Amines by Nb2o5 As a Reusable Lewis Acid Catalyst
Title Amidation of Carboxylic Acids with Amines by Nb2O5 as a Reusable Lewis Acid Catalyst Author(s) Ali, Md. A.; Siddiki, S. M. A. H.; Onodera, Wataru; Kon, Kenichi; Shimizu, Ken-ichi ChemCatChem, 7(21), 3555-3561 Citation https://doi.org/10.1002/cctc.201500672 Issue Date 2015-11-02 Doc URL http://hdl.handle.net/2115/63409 This is the accepted version of the following article: ChemCatChem. 7, 3555-3561(2015), which has been published in Rights final form at http://onlinelibrary.wiley.com/doi/10.1002/cctc.201500672. Type article (author version) File Information amide-wi.pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP Amidation of Carboxylic Acids with Amines by Nb2O5 as Reusable Lewis Acid Catalyst Md. A. Ali,[a] S. M. A. H. Siddiki,[b] Wataru Onodera,[a] Kenichi Kon,[a] Ken-ichi Shimizu*[a,b] [a] Catalysis Research Center, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan [b] Elements Strategy Initiative for Catalysts and Batteries, Kyoto University, Katsura, Kyoto 615-8520, Japan *Corresponding author Ken-ichi Shimizu Catalysis Research Center, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan E-mail: [email protected] Abstract: Among 28 types of heterogeneous and homogenous catalysts tested, Nb2O5 shows the highest yield for direct amidation of n-dodecanoic acid with a less reactive amine (aniline). The catalytic amidation by Nb2O5 is applicable to a wide range of carboxylic acids and amines with various functional groups, and the catalyst is reusable. Comparison of the results of the catalytic study and infrared (IR) study of the acetic acid adsorbed on the catalyst suggests that activation of carbonyl group of the carboxylic acid by Lewis acid sites on Nb2O5 is responsible for the high activity of Nb2O5. -

Alcohols and Ethers
ALCOHOLS AND ETHERS The hydroxyl group is one of the most important functional groups of na- turally occurring organic molecules. All carbohydrates and their derivatives, including nucleic acids, have hydroxyl groups. Some amino acids, most ste- roids, many terpenes, and plant pigments have hydroxyl groups. These sub- stances serve many diverse purposes for the support and maintenance of life. One extreme example is the potent toxin tetrodotoxin, which is isolated from puffer fish and has obvious use for defense against predators. This compound has special biochemical interest, having six different hydroxylic functions arranged on a cagelike structure: tetrodotoxin On the more practical side, vast quantities of simple alcohols - metha- nol, ethanol, 2-propanol, 1-butan01 - and many ethers are made from petro- leum-derived hydrocarbons. These alcohols are widely used as solvents and as intermediates for the synthesis of more complex substances. 15 Alcohols and Ethers The reactions involving the hydrogens of alcoholic OH groups are expected to be similar to those of water, HOH, the simplest hydroxylic com- pound. Alcohols, ROH, can be regarded in this respect as substitution products of water. However, with alcohols we shall be interested not only in reactions that proceed at the 8-H bond but also with processes that result in cleavage of the C-0 bond, or changes in the organic group R. The simple ethers, ROR, do not have 0-H bonds, and most of their reactions are limited to the substituent groups. The chemistry of ethers, there- fore, is less varied than that of alcohols. This fact is turned to advantage in the widespread use of ethers as solvents for a variety of organic reactions, as we already have seen for Grignard reagents (Section 14- 10). -

EDWARD C. KENDALL March 8, 1886-May 4, 1972
NATIONAL ACADEMY OF SCIENCES E D W A R D C . K ENDALL 1886—1972 A Biographical Memoir by Dw I G H T INGLE Any opinions expressed in this memoir are those of the author(s) and do not necessarily reflect the views of the National Academy of Sciences. Biographical Memoir COPYRIGHT 1975 NATIONAL ACADEMY OF SCIENCES WASHINGTON D.C. EDWARD C. KENDALL March 8, 1886-May 4, 1972 BY DWIGHT J. INGLE DWARD CALVIN KENDALL isolated thyroxine from the thyroid E gland; he and associates crystallized glutathione and estab- lished its chemical structure; and he and associates isolated a series of steroid compounds from the adrenal cortex and con- tributed importantly to the determination of the structure and synthesis of several of them. With Philip S. Hench, he con- ceived the idea that cortisone might be useful in treating rheumatoid arthritis, and they planned clinical studies that confirmed the hypothesis. Kendall also initiated and partici- pated in a number of related studies. During his professional life he was called "Nick" by close friends and by his wife. He was referred to as "The Chief" by some of his laboratory associates, but commonly he was ad- dressed with deference, as "Doctor Kendall." Edward C. Kendall, the third child of George S. and Eva F. Kendall, was born March 8, 1886, at South Norwalk, Connecti- cut. The home was a citadel for religious teachings. The father, a dentist by profession, took an active interest in com- munity affairs. Edward attended the Franklin Elementary School and, for two years, South Norwalk High School.