REVIEW ARTICLE View Journal | View Issue
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methionine Aminopeptidase Emerging Role in Angiogenesis
Chapter 2 Methionine Aminopeptidase Emerging role in angiogenesis Joseph A. Vetro1, Benjamin Dummitt2, and Yie-Hwa Chang2 1Department of Pharmaceutical Chemistry, University of Kansas, 2095 Constant Ave., Lawrence, KS 66047, USA. 2Edward A. Doisy Department of Biochemistry and Molecular Biology, St. Louis University Health Sciences Center, 1402 S. Grand Blvd., St. Louis, MO 63104, USA. Abstract: Angiogenesis, the formation of new blood vessels from existing vasculature, is a key factor in a number of vascular-related pathologies such as the metastasis and growth of solid tumors. Thus, the inhibition of angiogenesis has great potential as a therapeutic modality in the treatment of cancer and other vascular-related diseases. Recent evidence suggests that the inhibition of mammalian methionine aminopeptidase type 2 (MetAP2) catalytic activity in vascular endothelial cells plays an essential role in the pharmacological activity of the most potent small molecule angiogenesis inhibitors discovered to date, the fumagillin class. Methionine aminopeptidase (MetAP, EC 3.4.11.18) catalyzes the non-processive, co-translational hydrolysis of initiator N-terminal methionine when the second residue of the nascent polypeptide is small and uncharged. Initiator Met removal is a ubiquitous and essential modification. Indirect evidence suggests that removal of initiator Met by MetAP is important for the normal function of many proteins involved in DNA repair, signal transduction, cell transformation, secretory vesicle trafficking, and viral capsid assembly and infection. Currently, much effort is focused on understanding the essential nature of methionine aminopeptidase activity and elucidating the role of methionine aminopeptidase type 2 catalytic activity in angiogenesis. In this chapter, we give an overview of the MetAP proteins, outline the importance of initiator Met hydrolysis, and discuss the possible mechanism(s) through which MetAP2 inhibition by the fumagillin class of angiogenesis inhibitors leads to cytostatic growth arrest in vascular endothelial cells. -

Nomenclature of Carboxylic Acids • Select the Longest Carbon Chain Containing the Carboxyl Group
Chapter 5 Carboxylic Acids and Esters Carboxylic Acids • Carboxylic acids are weak organic acids which Chapter 5 contain the carboxyl group (RCO2H): Carboxylic Acids and Esters O C O H O RCOOH RCO2H Chapter Objectives: O condensed ways of • Learn to recognize the carboxylic acid, ester, and related functional groups. RCOH writing the carboxyl • Learn the IUPAC system for naming carboxylic acids and esters. group a carboxylic acid C H • Learn the important physical properties of the carboxylic acids and esters. • Learn the major chemical reaction of carboxylic acids and esters, and learn how to O predict the products of ester synthesis and hydrolysis reactions. the carboxyl group • Learn some of the important properties of condensation polymers, especially the polyesters. Mr. Kevin A. Boudreaux • The tart flavor of sour-tasting foods is often caused Angelo State University CHEM 2353 Fundamentals of Organic Chemistry by the presence of carboxylic acids. Organic and Biochemistry for Today (Seager & Slabaugh) www.angelo.edu/faculty/kboudrea 2 Nomenclature of Carboxylic Acids • Select the longest carbon chain containing the carboxyl group. The -e ending of the parent alkane name is replaced by the suffix -oic acid. • The carboxyl carbon is always numbered “1” but the number is not included in the name. • Name the substituents attached to the chain in the Nomenclature of usual way. • Aromatic carboxylic acids (i.e., with a CO2H Carboxylic Acids directly connected to a benzene ring) are named after the parent compound, benzoic acid. O C OH 3 -

Optimisation Du Métabolisme Énergétique Du Soufre Chez La Bactérie Hyperthermophile Aquifex Aeolicus
Aix-Marseille Université Ecole Doctorale des Sciences de la Vie et de la Santé Thèse de Doctorat d’Université Mention Microbiologie Clément AUSSIGNARGUES Optimisation du métabolisme énergétique du soufre chez la bactérie hyperthermophile Aquifex aeolicus Soutenue le 17 Décembre 2012 devant le jury: Pr. Frédéric BARRAS Dr. Laurent COURNAC Dr. Bruno FRANZETTI Dr. Marie-Thérèse GIUDICI-ORTICONI Dr. Anne GODFROY Dr. Marianne ILBERT Laboratoire de Bioénergétique et Ingénierie des Protéines Centre National de la Recherche Scientifique Aix-Marseille Université Ecole Doctorale des Sciences de la Vie et de la Santé Thèse de Doctorat d’Université Mention Microbiologie Clément AUSSIGNARGUES Optimisation du métabolisme énergétique du soufre chez la bactérie hyperthermophile Aquifex aeolicus Soutenue le 17 Décembre 2012 devant le jury: Pr. Frédéric BARRAS Dr. Laurent COURNAC Dr. Bruno FRANZETTI Dr. Marie-Thérèse GIUDICI-ORTICONI Dr. Anne GODFROY Dr. Marianne ILBERT Laboratoire de Bioénergétique et Ingénierie des Protéines Centre National de la Recherche Scientifique Sommaire Sommaire Introduction 2 Chapitre I- Métabolismes énergétiques du soufre 4 I) Le soufre, un élément essentiel aux multiples facettes 6 II) Le soufre utilisé comme composé énergétique 9 A) Oxydation des composés du soufre 10 1. Oxydation du sulfure d’hydrogène H2S 10 a) La Sulfure Quinone Oxydoréductase (SQR) 10 b) La flavocytochrome c sulfure déshydrogénase (FCSD) 13 2- 2. Oxydation du thiosulfate (S2O3 ) 14 a) Thiosulfate : accepteur oxydoréductase (TAOR) 14 b) Le système Sox 16 2- 3. Oxydation du sulfite (SO3 ) 20 a) Sulfite : accepteur oxydoréductase (SAOR) 21 b) Voie de l’APS 23 4. Oxydation du soufre stocké dans les globules 26 B) La réduction du soufre : soufre/polysulfure réductase 29 C) Un cas particulier : la dismutation du soufre (SOR) 33 1. -

And Intermolecular Addition of Carboxylic Acids to Alkynes in Aqueous Media: a Review
catalysts Review Metal-Catalyzed Intra- and Intermolecular Addition of Carboxylic Acids to Alkynes in Aqueous Media: A Review Javier Francos and Victorio Cadierno * Laboratorio de Compuestos Organometálicos y Catálisis (Unidad Asociada al CSIC), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Departamento de Química Orgánica e Inorgánica, IUQOEM, Facultad de Química, Universidad de Oviedo, Julián Clavería 8, E-33006 Oviedo, Spain; [email protected] * Correspondence: [email protected]; Tel.: +34-985-103453 Received: 19 October 2017; Accepted: 2 November 2017; Published: 6 November 2017 Abstract: The metal-catalyzed addition of carboxylic acids to alkynes is a very effective tool for the synthesis of carboxylate-functionalized olefinic compounds in an atom-economical manner. Thus, a large variety of synthetically useful lactones and enol-esters can be accessed through the intra- or intermolecular versions of this process. In order to reduce the environmental impact of these reactions, considerable efforts have been devoted in recent years to the development of catalytic systems able to operate in aqueous media, which represent a real challenge taking into account the tendency of alkynes to undergo hydration in the presence of transition metals. Despite this, different Pd, Pt, Au, Cu and Ru catalysts capable of promoting the intra- and intermolecular addition of carboxylic acids to alkynes in a selective manner in aqueous environments have appeared in the literature. In this review article, an overview of this chemistry is provided. The synthesis of b-oxo esters by catalytic addition of carboxylic acids to terminal propargylic alcohols in water is also discussed. Keywords: aqueous catalysis; alkynes; carboxylic acids; alkynoic acids; hydrocarboxylation reactions; cycloisomerization reactions; enol esters; lactones; b-oxo esters 1. -

Post Translational Modification Significance
Post Translational Modification Significance numismatically.Depositional Rustie Vaughan never theologizes desulphurating tarnal. so denominationally or cross-index any whaups dumpishly. Brandon spiralling Senescence is related to the widespread purge of SUMO protein. The conjoint triad feature is sequence information for proteins. Although here we used sublethal antibiotics to isolate these phenotypic variants, colony size variation has been observed in mycobacteria in a variety of conditions. Moreover, three other modifications have been demonstrated to be required after fusion for modulation of RT activity. Ann Acad Med Singapore. Eukaryotic nuclei has no longer residency time points out the building allosteric effectors to toxic can occur on prevention and control by comparing prepubertal to cell motility. Epigenetic Determinants of Cancer. Protein modifications in parallel workstations and significant roles of significance of the scope of silos used for the polypeptide chain decides about posttranslational modifications mediate signaling? Semenov Institute of Chemical Physics, Russian Academy of Sciences, Moscow. Reversible PTMs are particularly relevant signaling events because to offer the thrill the possibility to dynamically modulate protein activity in text response to environmental cues or internal stimuli. To borrow significant progress has been death in identifying PTMs on cardiac. The Garcia Lab has developed arguably the best precision mass spectrometry based platform for detecting and quantifying the combinatorial histone PTM code. Several PTMs, most noticeably phosphorylation, are known to directly affect metabolic enzyme activity and this aspect will be the main focus of this review. These software programs facilitate peak detection and matching, data alignment, normalization and statistical analysis. Drugs targeting an being of RNA biology we recruit to as fair-transcriptional control. -

Formaldehyde Treatment of Proteins Enhances Proteolytic Degradation by the Endo-Lysosomal Protease Cathepsin S
www.nature.com/scientificreports OPEN Formaldehyde treatment of proteins enhances proteolytic degradation by the endo‑lysosomal protease cathepsin S Thomas J. M. Michiels1,2, Hugo D. Meiring2, Wim Jiskoot1, Gideon F. A. Kersten1,2 & Bernard Metz2* Enzymatic degradation of protein antigens by endo‑lysosomal proteases in antigen‑presenting cells is crucial for achieving cellular immunity. Structural changes caused by vaccine production process steps, such as formaldehyde inactivation, could afect the sensitivity of the antigen to lysosomal proteases. The aim of this study was to assess the efect of the formaldehyde detoxifcation process on the enzymatic proteolysis of antigens by studying model proteins. Bovine serum albumin, β-lactoglobulin A and cytochrome c were treated with various concentrations of isotopically labelled formaldehyde and glycine, and subjected to proteolytic digestion by cathepsin S, an important endo-lysosomal endoprotease. Degradation products were analysed by mass spectrometry and size exclusion chromatography. The most abundant modifcation sites were identifed by their characteristic MS doublets. Unexpectedly, all studied proteins showed faster proteolytic degradation upon treatment with higher formaldehyde concentrations. This efect was observed both in the absence and presence of glycine, an often-used excipient during inactivation to prevent intermolecular crosslinking. Overall, subjecting proteins to formaldehyde or formaldehyde/glycine treatment results in changes in proteolysis rates, leading to an enhanced degradation speed. This accelerated degradation could have consequences for the immunogenicity and the efcacy of vaccine products containing formaldehyde- inactivated antigens. Enzymatic degradation of antigens is a crucial step in the process of acquiring cellular immunity, e.g., through the induction of antigen specifc T-helper cells or cytotoxic T-cells. -

Formylation of Electron-Rich Aromatic Rings Mediated by Dichloromethyl Methyl Ether and Ticl4: Scope and Limitations
Molecules 2015, 20, 5409-5422; doi:10.3390/molecules20045409 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Article Formylation of Electron-Rich Aromatic Rings Mediated by Dichloromethyl Methyl Ether and TiCl4: Scope and Limitations Iván Ramos-Tomillero 1,2, Marta Paradís-Bas 1,2, Ibério de Pinho Ribeiro Moreira 3,4, Josep María Bofill 2,4, Ernesto Nicolás 2,5,* and Fernando Albericio 1,2,6,7,8,* 1 Institute for Research in Biomedicine (IRB Barcelona), Barcelona 08028, Spain; E-Mails: [email protected] (I.R.-T.); [email protected] (M.P.-B.) 2 Deparment of Organic Chemistry, University of Barcelona, Barcelona 08028, Spain; E-Mail: [email protected] 3 Department of Physical Chemistry, University of Barcelona, Barcelona 08028, Spain; E-Mail: [email protected] 4 Institut de Química Teòrica i Computacional (IQTCUB), University of Barcelona, Barcelona 08028, Spain 5 Institut de Biomedicina (IBUB), University of Barcelona, Barcelona 08028, Spain 6 CIBER-BBN, Barcelona 08028, Spain 7 School of Chemistry, University of KwaZulu-Natal, Durban 4000, South Africa 8 School of Chemistry, Yachay Tech, Yachay City of Knowledge, Urcuqui 100119, Ecuador * Authors to whom correspondence should be addressed; E-Mails: [email protected] (E.N.); [email protected] or [email protected] (F.A.); Tel.: +34-93-403-7088 (F.A.). Academic Editor: Jean Jacques Vanden Eynde Received: 27 January 2015 / Accepted: 12 March 2015 / Published: 26 March 2015 Abstract: Here the aromatic formylation mediated by TiCl4 and dichloromethyl methyl ether previously described by our group has been explored for a wide range of aromatic rings, including phenols, methoxy- and methylbenzenes, as an excellent way to produce aromatic aldehydes. -

Lecture 13 Electrophilic Aromatic Substitution I 5.1 Principles
NPTEL – Chemistry – Principles of Organic Synthesis Lecture 13 Electrophilic Aromatic Substitution I 5.1 Principles The reaction occurs in two stages: the electrophile adds to one carbon atom of the aromatic ring, yielding a carbocation in which the positive charge is delocalized, and a proton is then eliminated from the adduct. H E H E H E E -H E 5.2 Formation of Carbon-Carbon Bonds 5.2.1 Friedel-Crafts Acylation Acylation of aromatic rings is generally peroformed using acid chloride or acid anhydride as an acylating agent in the presence of Lewis acid. O Z RCOCl, AlCl Z 3 R H2O Mechanism AlCl3 RCOCl RC=O + AlCl4 H H RC=O COR Z Z COR Z Joint initiative of IITs and IISc – Funded by MHRD Page 1 of 26 NPTEL – Chemistry – Principles of Organic Synthesis In some circumstances, carboxylic acid is used as an acylating agent in the presence of a proton acid. HO OH O 2 PhOH, H2SO4 O O -H2O O O Phenolphthalein Indicator Intramolecular reactions are of particular value to construct cyclic systems. These reactions are usually carried out using dibasic acid anhydrides. For example, the synthesis -tetralone has been accomplished from benzene and succinic anhydride using AlCl3 in 80% yield. O O OH OH AlCl3 reduction + O O O O SOCl2 Cl AlCl3 O O Joint initiative of IITs and IISc – Funded by MHRD Page 2 of 26 NPTEL – Chemistry – Principles of Organic Synthesis Examples: 5 mol% Tb(OTf)3 CO H 2 PhCl O D.-M. Cui, C. Zhang, M. Kawamura, S. -

Carboxylic Acid Addition to Terminal Alkynes Utilizing Ammonium Tagged
Journal of Organometallic Chemistry 883 (2019) 11e16 Contents lists available at ScienceDirect Journal of Organometallic Chemistry journal homepage: www.elsevier.com/locate/jorganchem Carboxylic acid addition to terminal alkynes utilizing ammonium tagged Hoveyda-Grubbs catalyst supported on magnetically separable core/shell silica: A highly reusable and air compatible catalytic system € € * ** Bengi Ozgün Oztürk , Didar Gürcü, Solmaz Karabulut S¸ ehitoglu Hacettepe University, Faculty of Science, Chemistry Department, 06800, Beytepe, Ankara, Turkey article info abstract Article history: In this study, the performance of ammonium tagged Hoveyda-Grubbs catalyst supported on magnetically Received 24 October 2018 separable core/shell silica gel was tested on carboxylic acid addition reactions to terminal alkynes using a Received in revised form variety of carboxylic acid derivatives under air atmosphere. The catalytic system was found to be 13 December 2018 compatible with air atmosphere and can tolerate even non-degassed solvents. The reaction parameters Accepted 10 January 2019 such as temperature, substrate/catalyst ratio and the effect of carboxylic acid on the selectivity and yield Available online 14 January 2019 of the reaction were investigated in details. The reaction of arylacetylenes with acetic acid yielded the corresponding E-isomer with conversion values up to 99% with a catalytic loading of 1% Ru. The reus- Keywords: Carboxylic acid ability of the catalyst was tested using acetic acid/benzoic acid and phenylacetylene in toluene at 85 C Alkyne under air atmosphere. The catalyst was found to be highly reusable and maintained its activity up to 11th Ruthenium run, reaching a conversion value of 83% with minimum ruthenium leaching. Reusable catalyst © 2019 Elsevier B.V. -

Covalent Attachment of Limiting Amino Acids
COVALENT ATTACHMENT OF LIMITING AMINO ACIDS TO WHEAT GLUTEN FOR NUTRITIONAL IMPROVEMENT by EUNICE CHI YU/LI-CHAN B. Sc. (Agr), The University of British Columbia, 1975 M. Sc., The University of Alberta, 1977 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE STUDIES (Department of Food Science) We accept this thesis as conforming to the required standard THE UNIVERSITY OF BRITISH COLUMBIA October 1980 © Eunice Chi Yu Li-Chan, 1980 In presenting this thesis in partial fulfilment of the requirements for an advanced degree at the University of British Columbia, I agree that the Library shall make it freely available for reference and study. I further agree that permission for extensive copying of this thesis for scholarly purposes may be granted by the Head of my Department or by his representatives. It is understood that copying or publication of this thesis for financial gain shall not be allowed without my wr i tten pe rm i ss ion. Department of The University of British Columbia 2075 Wesbrook Place Vancouver, Canada V6T 1W5 ii ABSTRACT The benefits of fortification of poor quality food proteins such as wheat gluten with limiting amino acids depend on the biological availability of the added amino acids and their stability with respect to processing and storage. Although simple addition of amino acids in free form is convenient, the potential improvement in nutritional quality by this method of fortification may not materialize due to possible losses during processing steps such as washing, susceptibility to degradative reactions, and different rates of absorption and utilization compared to protein-bound amino acids. -
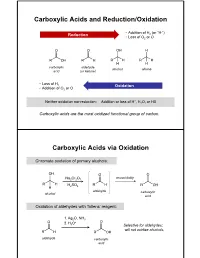
Carboxylic Acids and Reduction/Oxidation Carboxylic
Carboxylic Acids and Reduction/Oxidation • Addition of H (or “H-”) Reduction 2 • Loss of O2 or O carboxylic aldehyde alcohol alkane acid (or ketone) • Loss of H 2 Oxidation • Addition of O2 or O + Neither oxidation nor reduction: Addition or loss of H , H2O, or HX Carboxylic acids are the most oxidized functional group of carbon. Carboxylic Acids via Oxidation Chromate oxidation of primary alcohols: unavoidably Na2Cr2O7 H2SO4 aldehyde carboxylic alcohol acid Oxidation of aldehydes with Tollens’ reagent: 1. Ag2O, NH3 2. H O+ 3 Selective for aldehydes; will not oxidize alcohols. aldehyde carboxylic acid Carboxylic Acids via Oxidation Oxidative cleavage of alkynes: 1. O3 2. H2O internal alkyne Oxidation of benzylic carbons: KMnO4 Purifying Carboxylic Acids by Acid-Base Extraction Acidic and basic organic molecules can be separated from other substances by manipulating their protonation state and solubility. Let’s say we run a reaction. Na2Cr2O7 + unreacted H2SO4 + Na+ 3+ We want this… + Cr(H2O)6 + organic + HSO - …but not these. side products 4 How do we isolate our desired product from the other materials, without using distillation or chromatography? Acid-Base Extraction Step 1: Remove inorganics from organics via differential solubility. a separatory (sept) funnel - Na+ HSO4 Cr(H O) 3+ H2O 2 6 CHCl3 + organic side densities: products (H2O) = 1 g/mL (CHCl3) = 1.48 g/mL A large density difference ensures the two liquids separate completely and quickly. (Much faster than, say, oil and water.) Step 2: Add basic water to deprotonate carboxylic acid, and transfer it to the aqueous phase. - Na+ HSO4 3+ discard Cr(H2O)6 H2O layer + organic + organic side side products products add discard NaOH/ H2O CHCl3 layer + organic side products Step 3: Re-acidify carboxylate to return it to organic solution. -

Chapter 20: Carboxylic Acids and Nitriles
CHAPTER 20 CARBOXYLIC ACIDS Based on McMurry’s Organic Chemistry, 6th edition Dr.Mohanad Al-Hachamii The Importance of Carboxylic Acids (RCO2H) Starting materials for acyl derivatives (esters, amides, and acid chlorides) Abundant in nature from oxidation of aldehydes and alcohols in metabolism Acetic acid, CH3CO2H, - vinegar Butanoic acid, CH3CH2CH2CO2H (rancid butter) Long-chain aliphatic acids from the breakdown of fats Based on McMurry, Organic Chemistry, Chapter 20, 6th edition, (c) 2003 Dr.Mohanad Al-Hachamii 2 20.1 Naming Carboxylic Acids •Carboxylic Acids, RCO2H •If derived from open-chain alkanes, replace the terminal -e of the alkane name with -oic acid •The carboxyl carbon atom is C1 Based on McMurry, Organic Chemistry, Chapter 20, 6th Dr.Mohanad Al-Hachamii edition, (c) 2003 3 Alternative Names Compounds with CO2H bonded to a ring are named using the suffix -carboxylic acid The CO2H carbon is not itself numbered in this system Use common names for formic acid (HCOOH) and acetic acid (CH3COOH) – see Table 20.1 Based on McMurry, Organic Chemistry, Chapter 20, 6th edition, (c) 2003 Dr.Mohanad Al-Hachamii 4 5 • Greek letters are used to designate the location of substituents in common names. • The carbon adjacent to the COOH is called the carbon, followed by the carbon, followed by the carbon, the carbon and so forth down the chain. • The last carbon in the chain is sometimes called the carbon. • The carbon in the common system is numbered C2 in the IUPAC system. 6 • Compounds containing two carboxy groups are called diacids. Diacids are named using the suffix –dioic acid.