PORCN Inhibition Synergizes with PI3K/Mtor Inhibition in Wnt-Addicted Cancers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Influencers on Thyroid Cancer Onset: Molecular Genetic Basis
G C A T T A C G G C A T genes Review Influencers on Thyroid Cancer Onset: Molecular Genetic Basis Berta Luzón-Toro 1,2, Raquel María Fernández 1,2, Leticia Villalba-Benito 1,2, Ana Torroglosa 1,2, Guillermo Antiñolo 1,2 and Salud Borrego 1,2,* 1 Department of Maternofetal Medicine, Genetics and Reproduction, Institute of Biomedicine of Seville (IBIS), University Hospital Virgen del Rocío/CSIC/University of Seville, 41013 Seville, Spain; [email protected] (B.L.-T.); [email protected] (R.M.F.); [email protected] (L.V.-B.); [email protected] (A.T.); [email protected] (G.A.) 2 Centre for Biomedical Network Research on Rare Diseases (CIBERER), 41013 Seville, Spain * Correspondence: [email protected]; Tel.: +34-955-012641 Received: 3 September 2019; Accepted: 6 November 2019; Published: 8 November 2019 Abstract: Thyroid cancer, a cancerous tumor or growth located within the thyroid gland, is the most common endocrine cancer. It is one of the few cancers whereby incidence rates have increased in recent years. It occurs in all age groups, from children through to seniors. Most studies are focused on dissecting its genetic basis, since our current knowledge of the genetic background of the different forms of thyroid cancer is far from complete, which poses a challenge for diagnosis and prognosis of the disease. In this review, we describe prevailing advances and update our understanding of the molecular genetics of thyroid cancer, focusing on the main genes related with the pathology, including the different noncoding RNAs associated with the disease. -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

Mutational Landscape Differences Between Young-Onset and Older-Onset Breast Cancer Patients Nicole E
Mealey et al. BMC Cancer (2020) 20:212 https://doi.org/10.1186/s12885-020-6684-z RESEARCH ARTICLE Open Access Mutational landscape differences between young-onset and older-onset breast cancer patients Nicole E. Mealey1 , Dylan E. O’Sullivan2 , Joy Pader3 , Yibing Ruan3 , Edwin Wang4 , May Lynn Quan1,5,6 and Darren R. Brenner1,3,5* Abstract Background: The incidence of breast cancer among young women (aged ≤40 years) has increased in North America and Europe. Fewer than 10% of cases among young women are attributable to inherited BRCA1 or BRCA2 mutations, suggesting an important role for somatic mutations. This study investigated genomic differences between young- and older-onset breast tumours. Methods: In this study we characterized the mutational landscape of 89 young-onset breast tumours (≤40 years) and examined differences with 949 older-onset tumours (> 40 years) using data from The Cancer Genome Atlas. We examined mutated genes, mutational load, and types of mutations. We used complementary R packages “deconstructSigs” and “SomaticSignatures” to extract mutational signatures. A recursively partitioned mixture model was used to identify whether combinations of mutational signatures were related to age of onset. Results: Older patients had a higher proportion of mutations in PIK3CA, CDH1, and MAP3K1 genes, while young- onset patients had a higher proportion of mutations in GATA3 and CTNNB1. Mutational load was lower for young- onset tumours, and a higher proportion of these mutations were C > A mutations, but a lower proportion were C > T mutations compared to older-onset tumours. The most common mutational signatures identified in both age groups were signatures 1 and 3 from the COSMIC database. -

Ncomms3787.Pdf
ARTICLE Received 6 Aug 2013 | Accepted 17 Oct 2013 | Published 14 Nov 2013 DOI: 10.1038/ncomms3787 OPEN Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin Matthias Zebisch1, Yang Xu2,3, Christos Krastev1, Bryan T. MacDonald2, Maorong Chen2, Robert J.C. Gilbert1,XiHe2 & E. Yvonne Jones1 The four R-spondin (Rspo) proteins are secreted agonists of Wnt signalling in vertebrates, functioning in embryogenesis and adult stem cell biology. Through ubiquitination and degradation of Wnt receptors, the transmembrane E3 ubiquitin ligase ZNRF3 and related RNF43 antagonize Wnt signalling. Rspo ligands have been reported to inhibit the ligase activity through direct interaction with ZNRF3 and RNF43. Here we report multiple crystal structures of the ZNRF3 ectodomain (ZNRF3ecto), a signalling-competent Furin1–Furin2 (Fu1–Fu2) fragment of Rspo2 (Rspo2Fu1–Fu2), and Rspo2Fu1–Fu2 in complex with ZNRF3ecto,or RNF43ecto. A prominent loop in Fu1 clamps into equivalent grooves in the ZNRF3ecto and RNF43ecto surface. Rspo binding enhances dimerization of ZNRF3ecto but not of RNF43ecto. Comparison of the four Rspo proteins, mutants and chimeras in biophysical and cellular assays shows that their signalling potency depends on their ability to recruit ZNRF3 or RNF43 via Fu1 into a complex with LGR receptors, which interact with Rspo via Fu2. 1 Division of Structural Biology, Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford OX3 7BN, UK. 2 F.M. Kirby Neurobiology Center, Department of Neurology, Boston Children’s Hospital, Harvard Medical School, Boston, Massachusetts 02115, USA. 3 Key Laboratory for Molecular Enzymology and Engineering of Ministry of Education, College of Life Science, Jilin University, Changchun 130012, China. -

Molecular Mechanisms Underlying Innate Immune Kinase
MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION APPROVED BY SUPERVISORY COMMITTEE Michael A White, Ph.D. Melanie H. Cobb, Ph.D. Lawrence Lum, Ph.D. John D. Minna, M.D. DEDICATION This work is dedicated to my mother and Arlene for their love and support. ACKNOWLEDGEMENTS I am very grateful to my mentor, Dr. Michael White, for his continuous support and guidance through the entire study. I really appreciate his inspiration, patience, and generosity. I would also like to thank my committee members, Dr. Cobb, Dr. Lum, and Dr. Minna, for their invaluable advice and discussion. I thank all the White lab members and my friends for their help, suggestion, and discussion. Particularly, I would like to thank Rosie, Michael, Brian, Tzuling, and Malia for their long-term support and collaboration. I would also like to thank my friends, Veleka, Pei-Ling, Jen-Chieh, Shu-Yi, Chih-Chiang, Jen-Shuan, Yi-Chun, and Yu-San for their friendship. I am grateful to Drs. Rolf Brekken, Zhijian James Chen, Xuetao Cao, Philip Tsichlis, Charles Yeaman, William Hahn, Keqiang Ye, and Shu-Chan Hsu, Bing Su, Dos Sarbassov, Mark Magnuson, David Sabatini, Thomas Tan, and Bert Vogelstein for many of the reagents used in these studies. Finally, and most importantly, I would like to thank my mother and Arlene for their unending support and encouragement. MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION by YI-HUNG OU DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas April, 2013 Copyright by YI-HUNG OU, 2013 All Rights Reserved MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION Publication No. -
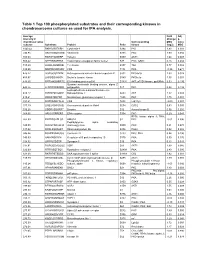
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Members of the Competence Network for Congenital Heart Defects, Germany
Members of the Competence Network for Congenital Heart Defects, Germany Hashim Abdul-Khaliq, Hans-Heiner Kramer, Felix Berger, Brigitte Stiller, Ulrike Bauer, Thomas Pickardt, Sabine Klaassen Family 49 Family 62 Family 226 Family 333 * * * * * * * * TOF AVS ASD ASD PAPVR TOF * * * * ASD PAPVD COA ASD * Family 346 Family 398 VSD * Family 489 * * VSD VSD * * VSD HCM * * * Family 545 BAV AVS ASD PDA Sv AS VSD Family 576 Family 645 Septal defect * * * COA AVS ASD BAV VSD * * * * BAV AVS AVS ASD BAV Family 702 * VSD VSD Family 732 * Family 720 AVSD * * * VSD * * Family 831 TOF TOF TOF Vring TGA VSD * * * * PDA VSD AVS * PDA * * * VSD BAV PFO ASD ASD2 PDA PDA COA Family 1117 Family 1121 * PVS ? * ASD VSD * * * TOF VSD ASD VSD Family 1319 Family 1151 * * * * 2 * ASD ASD * * ASD ASD * * EbA AVS AVS Family 1364 Family 1560 Family 1575 * * TOF VSD ASD VSD infPS TOF * * * VSD PVS PVS PVS VSD * VSD PVS Family 1710 ASD Family 1722 * ASD CHD * VSD VSD * COA TGA DCM ASD BAV SV, MVA COA PVS * DCM DCM ASD HLHS COA BAV Family 2077 Family 2261 * COA, BAV * * * PVS PVS PVS * * infPS infPS BAV ASD * * AVS ASD ASD BAV BAV Family 3500 Family 2558 Family 3315 * * * AVS VSD AVR * * HLHS COA PDA ASD BAV * PVA Family 3501 VSD Family 3503 ? ? * * * VSD HRHS EbA * 3 PVA PAA ASD PDA VSD Family 3505 Family 3540 * * BAV ASD AVS * * * HLHS HLHS TAPVR BAV COA Figure S1. Pedigrees of 32 Danish multiplex CHD families. Circles: females. Squares: males. White symbols: unaffected family members. Filled symbols: affected family members. Triangles: abortion. -

Inhibition of ERK 1/2 Kinases Prevents Tendon Matrix Breakdown Ulrich Blache1,2,3, Stefania L
www.nature.com/scientificreports OPEN Inhibition of ERK 1/2 kinases prevents tendon matrix breakdown Ulrich Blache1,2,3, Stefania L. Wunderli1,2,3, Amro A. Hussien1,2, Tino Stauber1,2, Gabriel Flückiger1,2, Maja Bollhalder1,2, Barbara Niederöst1,2, Sandro F. Fucentese1 & Jess G. Snedeker1,2* Tendon extracellular matrix (ECM) mechanical unloading results in tissue degradation and breakdown, with niche-dependent cellular stress directing proteolytic degradation of tendon. Here, we show that the extracellular-signal regulated kinase (ERK) pathway is central in tendon degradation of load-deprived tissue explants. We show that ERK 1/2 are highly phosphorylated in mechanically unloaded tendon fascicles in a vascular niche-dependent manner. Pharmacological inhibition of ERK 1/2 abolishes the induction of ECM catabolic gene expression (MMPs) and fully prevents loss of mechanical properties. Moreover, ERK 1/2 inhibition in unloaded tendon fascicles suppresses features of pathological tissue remodeling such as collagen type 3 matrix switch and the induction of the pro-fbrotic cytokine interleukin 11. This work demonstrates ERK signaling as a central checkpoint to trigger tendon matrix degradation and remodeling using load-deprived tissue explants. Tendon is a musculoskeletal tissue that transmits muscle force to bone. To accomplish its biomechanical function, tendon tissues adopt a specialized extracellular matrix (ECM) structure1. Te load-bearing tendon compart- ment consists of highly aligned collagen-rich fascicles that are interspersed with tendon stromal cells. Tendon is a mechanosensitive tissue whereby physiological mechanical loading is vital for maintaining tendon archi- tecture and homeostasis2. Mechanical unloading of the tissue, for instance following tendon rupture or more localized micro trauma, leads to proteolytic breakdown of the tissue with severe deterioration of both structural and mechanical properties3–5. -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -

Multi Gene Mutation Signatures in Colorectal Cancer
Zhuang et al. BMC Cancer (2021) 21:380 https://doi.org/10.1186/s12885-021-08108-9 RESEARCH ARTICLE Open Access Multi gene mutation signatures in colorectal cancer patients: predict for the diagnosis, pathological classification, staging and prognosis Yan Zhuang1†, Hailong Wang2†, Da Jiang3, Ying Li3, Lixia Feng4, Caijuan Tian5, Mingyu Pu5, Xiaowei Wang6, Jiangyan Zhang6, Yuanjing Hu7* and Pengfei Liu2* Abstract Background: Identifying gene mutation signatures will enable a better understanding for the occurrence and development of colorectal cancer (CRC), and provide some potential biomarkers for clinical practice. Currently, however, there is still few effective biomarkers for early diagnosis and prognostic judgment in CRC patients. The purpose was to identify novel mutation signatures for the diagnosis and prognosis of CRC. Methods: Clinical information of 531 CRC patients and their sequencing data were downloaded from TCGA database (training group), and 53 clinical patients were collected and sequenced with targeted next generation sequencing (NGS) technology (validation group). The relationship between the mutation genes and the diagnosis, pathological type, stage and prognosis of CRC were compared to construct signatures for CRC, and then analyzed their relationship with RNA expression, immunocyte infiltration and tumor microenvironment (TME). (Continued on next page) * Correspondence: [email protected]; [email protected] †Yan Zhuang and Hailong Wang contributed equally to this work. 7Department of Gynecological Oncology, Tianjin Central Hospital of Obstetrics & Gynecology, No. 156 Nankai Third Road, Nankai District, Tianjin 300100, China 2Department of Oncology, Tianjin Academy of Traditional Chinese Medicine Affiliated Hospital, No.354 Beima Road, Hongqiao District, Tianjin 300120, China Full list of author information is available at the end of the article © The Author(s). -

The Curing AI for Precision Medicine
The Curing AI for Precision Medicine Hoifung Poon 1 Medicine Today Is Imprecise Top 20 drugs 80% non-responders Wasted 1/3 health spending $750 billion / year 2 Disruption 1: Big Data 2009 2013: 40% 93% 3 Disruption 2: Pay-for-Performance Goal: 75% by 2020 4 Vemurafenib on BRAF-V600 Melanoma Before Treatment 15 Weeks 5 Vemurafenib on BRAF-V600 Melanoma Before Treatment 15 Weeks 23 Weeks 6 Why We Haven’t Solved Precision Medicine? … ATTCGGATATTTAAGGC … … ATTCGGGTATTTAAGCC … … ATTCGGATATTTAAGGC … … ATTCGGGTATTTAAGCC … … ATTCGGATATTTAAGGC … … ATTCGGGTATTTAAGCC … High-Throughput Data Discovery Bottleneck #1: Knowledge Bottleneck #2: Reasoning AI is the key to overcome these bottlenecks 7 Use Case: Molecular Tumor Board 8 www.ucsf.edu/news/2014/11/120451/bridging-gap-precision-medicine Use Case: Molecular Tumor Board Problem: Hard to scale U.S. 2015: 1.6 million new cases, 600K deaths 902 cancer hospitals Memorial Sloan Kettering 2016: Sequence: Tens of thousand Board can review: A few hundred Wanted: Decision support for cancer precision medicine 9 First-Generation Molecular Tumor Board Knowledge bottleneck E.g., given a tumor sequence, determine: What genes and mutations are important What drugs might be applicable Can do manually but hard to scale 10 Next-Generation Molecular Tumor Board Reasoning bottleneck E.g., personalize drug combinations Can’t do manually, ever 11 Big Medical Data Decision Support Precision Medicine Machine Predict Reading Drug Combo 12 13 PubMed 26 millions abstracts Two new abstracts every minute Adds over one million every year 14 Machine Reading PMID: 123 … VDR+ binds to SMAD3 to form … PMID: 456 Knowledge … JUN expression Base is induced by SMAD3/4 … …… 15 Machine Reading Involvement of p70(S6)-kinase activation in IL-10 up-regulation in human monocytes by gp41 envelope protein of human immunodeficiency virus type 1 ..