Synthesis of New Functionalized Organocopper Reagents Via a Halogen-Copper Exchange Reaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FAROOK COLLEGE (Autonomous)
FAROOK COLLEGE (Autonomous) M.Sc. DEGREE PROGRAMME IN CHEMISTRY CHOICE BASED CREDIT AND SEMESTER SYSTEM-PG (FCCBCSSPG-2019) SCHEME AND SYLLABI 2019 ADMISSION ONWARDS 1 CERTIFICATE I hereby certify that the documents attached are the bona fide copies of the syllabus of M.Sc. Chemistry Programme to be effective from the academic year 2019-20 onwards. Date: Place: P R I N C I P A L 2 FAROOK COLLEGE (AUTONOMOUS) MSc. CHEMISTRY (CSS PATTERN) Regulations and Syllabus with effect from 2019 admission Pattern of the Programme a. The name of the programme shall be M.Sc. Chemistry under CSS pattern. b. The programme shall be offered in four semesters within a period of two academic years. c. Eligibility for admission will be as per the rules laid down by the College from time to time. d. Details of the courses offered for the programme are given in Table 1. The programme shall be conducted in accordance with the programme pattern, scheme of examination and syllabus prescribed. Of the 25 hours per week, 13 hours shall be allotted for theory and 12 hours for practical; 1 theory hour per week during even semesters shall be allotted for seminar. Theory Courses In the first three semesters, there will be four theory courses; and in the fourth semester, three theory courses. All the theory courses in the first and second semesters are core courses. In the third semester there will be three core theory courses and one elective theory course. College can choose any one of the elective courses given in Table 1. -

Organometallic Compounds
Chapter 11 Organometallic compounds Organometallics Reactions using organometallics Organometallic compounds Ch 11 #2 = comp’s containing a carbon-metal bond δ− δ+ C in organometallic comp’ds are nucleophilic. C in organic comp’d (like ROH, RNH , and RX) 2 δ+ δ are electrophilic. − due to ∆EN Ch 11 #3 in substitution reactions a carbon Nu: R-Li and R-MgX Ch 11 #4 (used to be) the two most common organometallics organolithium comp’ds BuLi = an alkyl lithium organomagnesium comp’ds = Grignard reagents 1912 Nobel Prize R, Ar, vinyl all possible; Br (as X) popular Ether (solvent) coordinates Mg, stabilizing it. Reactions of R-Li and R-MgX Ch 11 #5 reacts like a carbanion C:– ~ a C Nu: reactions as C Nu: (like SN(2)) nucleophilic addition to carbonyls ~ more often Chapt 16 Ch 11 #6 R-Li and R-MgX are very strong B: how strong? pKa? − − react even with very weak acid stronger than OH? NH2? useful for preparing deuterated HC Storage and reaction must be acid- and moisture-free. Transmetal(l)ation Ch 11 #7 R-Li is more reactive than R-MgX is. C–Li more polar than C–Mg C of R-Li more nu-philic [better Nu:] transmetalation [metal exchange] to less reactive [more stable] organometallic Coupling using Gilman reagent Ch 11 #8 coupling reaction (in organic chemistry) two hydrocarbon fragments are coupled (to form C–C) with the aid of a (transition) metal catalyst Gilman reagent coupling of R of R-X and R’ of Gilman reagent RX + R’2CuLi R–R’ mechanism? substitution of X with R’? not clear Ch 11 #9 R can be alkyl, aryl, or alkenyl [vinyl] which is not possible by R-Li or R-MgX Is R of Gilman why? they are SN(2). -
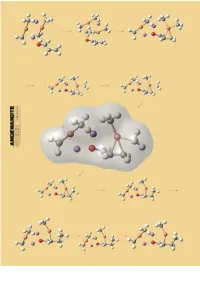
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

Organocuprate Aggregation and Reactivity: Decoding the 'Black Box'
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München Organocuprate Aggregation and Reactivity: Decoding the ‘Black Box’ Aliaksei Putau aus Vitebsk, Weißrussland 2012 Erklärung Diese Dissertation wurde im Sinne von § 7 der Promotionsordnung vom 28. November 2011 von Herrn Prof. Dr. Konrad Koszinowski betreut, und von Herrn Prof. Dr. Mayr von der Fakultät für Chemie und Pharmazie vertreten. Eidesstattliche Versicherung Diese Dissertation wurde selbständig, ohne unerlaubte Hilfe erarbeitet. München, 11.06.2012 _______________________ Aliaksei Putau Dissertation eingereicht am: 12.06.2012 1. Gutachter Prof. Dr. Herbert Mayr 2. Gutachter Prof. Dr. Konrad Koszinowski Mündliche Prüfung am: 20.07.2012 Acknowledgements It is a pleasure to thank the many people who made this project possible. My gratitude to my PhD supervisor, Prof. Dr. Konrad Koszinowski, is difficult to overstate. His sound advice, good company, and enthusiasm for trying new things ensured the tropical sunny climate in our group that is so necessary for fruitful (and, above all, enjoyable) research. Besides, his involvement in the group life outside the lab made all of us group members feel like team members, not just a loose bunch of colleagues. I am also deeply indebted to Prof. Dr. Herbert Mayr, not only for his continuous generous support and help, but also for running such a cool, all-star research group, which provided a great many social occasions. I thank SFB 749 for the financial support of this project, specifically Mrs. Birgit Carell, for her support and understanding of my worries and troubles of all kinds. Furthermore, my sincere gratitude goes to Prof. -

Select Reactions of Organoboranes and Organostannanes
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 8-2012 Select Reactions of Organoboranes and Organostannanes David W. Blevins [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Organic Chemistry Commons Recommended Citation Blevins, David W., "Select Reactions of Organoboranes and Organostannanes. " PhD diss., University of Tennessee, 2012. https://trace.tennessee.edu/utk_graddiss/1412 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by David W. Blevins entitled "Select Reactions of Organoboranes and Organostannanes." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Chemistry. George W. Kabalka, Major Professor We have read this dissertation and recommend its acceptance: Shane Foister, Craig Barnes, Kimberly Gwinn Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Select Reactions of Organoboranes and Organostannanes A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville David W. Blevins August 2012 DEDICATION I dedicate this work to Lisa, Preston, and Clarissa Blevins. ii ACKNOWLEDGEMENTS I would like to thank Dr. -

Chem 341 • Organic Chemistry I Lecture Summary 29 • November 02, 2007
Chem 341 • Organic Chemistry I Lecture Summary 29 • November 02, 2007 Chapter 10 - Alkyl Halides Preparation of Organometallic Compounds - Grignard reagents Alkyl halides are good electrophiles. We will examine their reactivity in nucleophilic substitution in detail in the next chapter. Another useful property of alkyl halides as that reducing metals can insert between the C-X bond to prepare a C-metal bond. This drastically alters the reactivity as the metal is less electronegative than carbon. Thus, the reactivity of the carbon is reversed from being an electrophile to being a nucleophile. Grignard reagents are formed from the insertion of Mg metal into the C-X bond. These reagents are very reactive as bases and electrophiles and are extremely sensitive to any source of proton (water, ROH, RNH2, etc). This can be useful if one wants to reduce an alkyl halide to an alkane. It is also a useful method of introducing hydrogen isotopes, such as deuterium, by reaction of the Grignard reagent with heavy water (D stands for Deuterium, the isotope of hydrogen 2H). Grignard reagents can be prepared from a variety of different kinds of organohalides (eg. Alkyl halides, vinyl halides, etc.). One important reaction of Grignard reagents is their ability to add to carbonyl compounds (C=O double bonds). You will study this in detail next semester. δ− δ+ δ+ Br δ− Mg Br D Mg D2O O reactivity of OH carbon reversed R H then H+ R Preparation of Organometallic Compounds - Gilman reagents Lithium metal reacts with organohalides in a simlar fashion as magnesium. Organolithium reagents are very basic and are often used to deprotonated compounds that are not very acidic. -

REVIEW Methyl Complexes of the Transition Metals
REVIEW Methyl Complexes of the Transition Metals Jesús Campos,[b] Joaquín López-Serrano,[a] Riccardo Peloso,[a] and Ernesto Carmona*[a] Dedicated to Prof. Pierre Braunstein in recognition of his contributions to inorganic and organometallic chemistry [a] Title(s), Initial(s), Surname(s) of Author(s) including Corresponding Author(s) Department Institution Address 1 E-mail: [b] Title(s), Initial(s), Surname(s) of Author(s) Department Institution Address 2 Supporting information for this article is given via a link at the end of the document.((Please delete this text if not appropriate)) REVIEW Abstract: Organometallic chemistry can be considered as a wide focusing in the last 5 years. It begins with a brief description of area of knowledge that combines concepts of classic organic synthetic methods, followed by a selection of recently reported chemistry, i.e. based essentially on carbon, with molecular inorganic complexes with terminal and bridging methyl ligands from the chemistry, especially with coordination compounds. Transition metal groups 3 to 11 of the periodic table. A specific section is methyl complexes probably represent the simplest and most dedicated to methyl-bridged species with three-centre two- fundamental way to view how these two major areas of chemistry electron bonds. The review concludes highlighting relevant combine and merge into novel species with intriguing features in reactivity of the methyl group in this class of compounds. terms of reactivity, structure, and bonding. Citing more than 500 bibliographic references, this review aims to offer a concise view of recent advances in the field of transition metal complexes containing Ernesto Carmona (PhD degree, University M—CH3 fragments. -

Chemistry for Pharmacy Students : General, Organic
Chemistry for Pharmacy Students General, Organic and Natural Product Chemistry Satyajit D. Sarker University of Ulster, Coleraine, Northern Ireland, UK Lutfun Nahar University of Ulster, Coleraine, Northern Ireland, UK Chemistry for Pharmacy Students Chemistry for Pharmacy Students General, Organic and Natural Product Chemistry Satyajit D. Sarker University of Ulster, Coleraine, Northern Ireland, UK Lutfun Nahar University of Ulster, Coleraine, Northern Ireland, UK Copyright #2007 John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England Telephone (þ44) 1243 779777 Email (for orders and customer service enquiries): [email protected] Visit our Home Page on www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1T 4LP, UK, without the permission in writing of the Publisher. Requests to the Publisher should be addressed to the Permissions Department, John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England, or emailed to [email protected], or faxed to (þ44) 1243 770620. Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The Publisher is not associated with any product or vendor mentioned in this book. -

Organometallic Chemistry — for Organic Synthesis Abbreviations in Chemical Structures
Organometallic Chemistry — for organic synthesis Abbreviations in Chemical Structures Me CH - methyl Bz PhC(O)- benzoyl Contents 3 Et MeCH2- ethyl Boc t-BuOCO tert-butoxycarbonyl 1. Main group organometallic chemistry Pr EtCH2- n-propyl Ts p-TolSO 2- p-toluenesulfonyl 1-1. Preparations Bu PrCH2- n-butyl Ms MeSO2- methanesulfonyl 1-2. Reactions Pent BuCH2- n-pentyl Tf CF3SO2- trifluoromethanesulfonyl Hex PentCH - n-hexyl TMS Me Si- trimethylsilyl 2. Reactions of organic molecules and transition-metal complexes 2 3 i-Pr Me2CH- isopropyl TES Et3Si- triethylsilyl 3. Reactions catalyzed by transition-metal complexes i-Bu i-PrCH- isobutyl TBS t-BuMe2Si- tert-butyldimethylsilyl 3-1. Hydrogenation s-Bu EtMeCH- sec-butyl 3-2. Cross-coupling and related reactions t-Bu Me3C- tert-butyl c-Pent c-C H - cyclopentyl 3-3. Olefin metathesis 5 9 t-Am EtMe2C- tert-amyl 3-4. Homogeneous metal catalysts in chemical industry Cy (c-Hex) c-C6H11- cyclohexyl Ph C6H5- phenyl pin -OCMe2CMe2O- pinacolate Bn PhCH2- benzyl cat 1,2-C6H4O2 catecholate Books o-Tol 2-MeC6H4- 2-methylphenyl 1. Comprehensive Organometallic Chemistry III m-Tol 3-MeC6H4- 3-methylphenyl R any C substituents eds. by D. M. P. Mingos and R. H. Crabtree, Elsevier, Oxford, 2007. p-Anis 4-MeOC6H4- 4-methoxyphenyl Ar any aromatic substituents (https://www.sciencedirect.com/science/referenceworks/9780080450476) PMB p-AnisCH2- 4-methoxybenzyl X any halogen and related leaving group Xyl 5-Me-m-Tol - 3,5-dimethylphenyl M any metal substituents 2. 第5版 実験化学講座 有機化合物の合成 VI 金属を用いる有機合成 Mes 2,4,6-Me3C6H2- mesityl 日本化学会 編, 丸善, 2004. -

Arts, Commerce & Science College, Bodwad
The Bodwad Sarvjanik Co-Op. Education Society Ltd., Bodwad Arts, Commerce & Science College, Bodwad. Question Bank Class: S.Y.B.Sc. Sem: IV Subject: Organic & Inorganic Chemistry Paper Name: Chemistry-II 1. Which of the following compounds is an example of active methylene compound? A. Malonic ester B. Ethyl alcohol C. Propane D. Acetone 2. Ethyl acetate on heating with sodium ethoxide gives….. A. Ethyl aceto acetate B. Sodium acetate C. Ethyl alcohol D. Diethyl ether 3. Ethylaceto acetate undergoes acid hydrolysis with dil HCl to give_____ A. Aceto acetic acid B. Succinic acid C. Acetic acid D. Adipic acid 4. Aceto acetic acid on decarboxylation gives ______ A. Ethanol B. Acetone C. Butanone D. Methanol 5. Base catalysed condensation of two ester molecules to form an alcohol and -keto ester is called A. Claisen Condensation B. Aldol condensation C. Cannizzaro reaction D. Benzoin condensation. 6. The base catalysed reaction of ethyl aceto acetate with an aldehyde or a ketone followed by acid hydrolysis gives A. alkyl acetic acid B. Dialkyl acetic acid C. , unsaturated acid D. ketone 7. The reaction of two moles of diethyl malonate with 1, 2 di iodo ethane followed by hydrolysis and decarboxylation gives_____ A. Succinic acid B. Glutaric acid C. Adipic acid D. None of these 8. Which of the following is necessary for the synthesis of diethyl malonate A. Acetic acid B. Potassium cyano acetate C. Ethyl acetate D. Methyl acetate 9. The acidity of hydrogen atoms of active methylene group is due to A. The resonance stabilization of the resultant carbanion B. -

Alkyl Halides
Alkyl Halides Alkyl Halides (RX)—compounds with a halogen bonded to a saturated carbon (sp3 hybridized carbon). Nomenclature Alkyl halides are named in the same way as alkanes. Treat the halogen as a substituent on the alkane parent chain. Example Cl 2-chloropentane Reactions to Know Synthesis of Alkyl Halides You learned in chapter 5 how to make an alkyl halide by a radical reaction between a halogen and an alkane in uv light. 1. From Alkenes by Allylic Bromination: H Br C C NBS C C C C Allylic Bromination with NBS is analogous to the radical reaction with an alkane, a halogen and uv light (Ch. 5). The NBS can be thought of as producing a Br radical. The Br radical removes a hydrogen, leaving an allylic radical and forming HBr. This allylic radical reacts with Br2 (which is formed from NBS reacting with the HBr formed from the removal of a hydrogen from the alkene) to give the product. H C C C C C Br C HBr Br2 Br C C C Br Why does the reaction only occur at the allylic position? Allylic radicals are more stable than alkyl or vinylic radicals: Stability of Radicals H H H R H C R C R C R C CC C CC H H R R Vinylic < methyl < primary < secondary < tertiary < allylic Allylic radicals are so stable because they are resonance stabilized. Because the allylic radical is more stable, it has a lower energy of activation and more collisions per unit of time result in a successful reaction at the allylic position. -

Synthetic Organic Chemistry Mscch-07 Vardhman Mahaveer Open University, Kota
MScCH-07 Vardhman Mahaveer Open University, Kota Synthetic Organic Chemistry MScCH-07 Vardhman Mahaveer Open University, Kota Synthetic Organic Chemistry Course Development Committee Chair Person Prof. Ashok Sharma Vice-Chancellor Vardhman Mahaveer Open University, Kota Coordinator and Members Coordinator SANDEEP HOODA Assistant Professor of Zoology School of Science & Technology Vardhman Mahaveer Open University, Kota Prof L.R. Gurjar Dr. Arvind Pareek Director Academic Director Regional Centre-Bharatpur VMOU Kota VMOU Kota Dr. Anuradha Dubey Dr. Sunil kumar Jangir Deputy Director,SOST Convener Chemistry VMOU Kota VMOU Kota Prof. P.S. Verma (Retd.) Prof. Pahup Singh (Retd.) Department of Chemistry Department of Chemistry University of Raj, Jaipur University of Raj, Jaipur Prof. P.D. Sharma (Retd.) Prof. Ashu Rani Department of Chemistry Department of Chemistry University of Raj, Jaipur University of Kota, Kota Dr. R.L. Pilaliya, (Retd.) Dr. Sapna Sharma Department of Chemistry, Govt. College Department of Chemistry Bikaner JECRC,university Jaipur Dr. Sanjay Kumar Sharma Dr. Sushil Kumar Sharma Department of Chemistry Department of Chemistry JECRC,university Jaipur University of Kota, Kota Editing and Course Writing Editor Prof. P.S. Verma (Retd.) Dr. Sushil Kumar Sharma Department of Chemistry Assistant Professor, Department of Pure University of Raj, Jaipur and Applied Chemistry University of Kota, Kota Writers: Dr. Shobhana Sharma 1,2,3,6 Dr.Sushil Kr. Sharma 4 ,8,11,14, Assistant Professor, Assistant Professor, Department of Chemistry Department of Pure and S.S.Jain Subodh Applied Chemistry P.G.College,Jaipur University of Kota, Kota Mr. Ankit Sharma 5.7,12 Dr.Girja Shankar Tailor 9,10 Assistant Professor, Assistant Professor, Department of Pure and Department of Chemistry Applied Chemistry Poddar University of Kota, Kota International College, Mansarovar, Jaipur Dr.