Synthetic Organic Chemistry Mscch-07 Vardhman Mahaveer Open University, Kota
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

And C,N-Chelated Organocopper Compounds†
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 2 September 2021 Review C,C- and C,N-Chelated Organocopper Compounds† Liang Liu1, Hui Chen2, Zhenqiang Yang2, Junnian Wei1* and Zhenfeng Xi1,3* 1 Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China; [email protected] (L.L.), [email protected] (J.W.), [email protected] (Z.X.) 2 Henan Institute of Chemistry Co. Ltd., Henan Academy of Sciences, Zhengzhou 450002, China; [email protected] (H.C.), [email protected] (Z.Y.) 3 State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Shanghai 200032, China; [email protected] (Z.X.) * Correspondence: [email protected] (J.W.), [email protected] (Z.X.) † Dedicated to Professor Gerard van Koten on the occasion of his 80th birthday Abstract: Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure- function relationship of organocopper compounds would advance rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono- carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize the unstable organocopper compounds. The bidentate ligands can chelate to the same copper atom via 휂2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via 휂1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. -

Antifouling Coating Composition, Coating Film Therefrom, Underwater Material Covered with the Coating Film and Antifouling Method
Europäisches Patentamt *EP001342756A1* (19) European Patent Office Office européen des brevets (11) EP 1 342 756 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C09D 5/16 10.09.2003 Bulletin 2003/37 (21) Application number: 03251373.1 (22) Date of filing: 06.03.2003 (84) Designated Contracting States: • Nakamura, Naoya AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Ohtake-shi, Hiroshima (JP) HU IE IT LI LU MC NL PT RO SE SI SK TR • Tsuboi, Makoto Designated Extension States: Ohtake-shi, Hiroshima (JP) AL LT LV MK (74) Representative: Cresswell, Thomas Anthony (30) Priority: 06.03.2002 JP 2002060696 J.A. KEMP & CO. 14 South Square (71) Applicant: CHUGOKU MARINE PAINTS, LTD. Gray’s Inn Ohtake-shi, Hiroshima (JP) London WC1R 5JJ (GB) (72) Inventors: • Oya, Masaaki, Ohtake-shi, Hiroshima (JP) (54) Antifouling coating composition, coating film therefrom, underwater material covered with the coating film and antifouling method (57) An antifouling coating composition comprising: (D) a dehydrating agent. (A) a silyl ester copolymer containing constituent units derived from a polymerizable unsaturated car- boxylic acid silyl ester, (B) a carboxylic acid, (C) a bivalent or trivalent metal compound, and EP 1 342 756 A1 Printed by Jouve, 75001 PARIS (FR) EP 1 342 756 A1 Description FIELD OF THE INVENTION 5 [0001] The present invention relates to an antifouling coating composition which contains a silyl ester copolymer, an antifouling coating film formed from the antifouling coating composition, an antifouling method wherein the antifouling coating composition is used, and a marine vessel (hull) or underwater structure covered with the coating film. -

FAROOK COLLEGE (Autonomous)
FAROOK COLLEGE (Autonomous) M.Sc. DEGREE PROGRAMME IN CHEMISTRY CHOICE BASED CREDIT AND SEMESTER SYSTEM-PG (FCCBCSSPG-2019) SCHEME AND SYLLABI 2019 ADMISSION ONWARDS 1 CERTIFICATE I hereby certify that the documents attached are the bona fide copies of the syllabus of M.Sc. Chemistry Programme to be effective from the academic year 2019-20 onwards. Date: Place: P R I N C I P A L 2 FAROOK COLLEGE (AUTONOMOUS) MSc. CHEMISTRY (CSS PATTERN) Regulations and Syllabus with effect from 2019 admission Pattern of the Programme a. The name of the programme shall be M.Sc. Chemistry under CSS pattern. b. The programme shall be offered in four semesters within a period of two academic years. c. Eligibility for admission will be as per the rules laid down by the College from time to time. d. Details of the courses offered for the programme are given in Table 1. The programme shall be conducted in accordance with the programme pattern, scheme of examination and syllabus prescribed. Of the 25 hours per week, 13 hours shall be allotted for theory and 12 hours for practical; 1 theory hour per week during even semesters shall be allotted for seminar. Theory Courses In the first three semesters, there will be four theory courses; and in the fourth semester, three theory courses. All the theory courses in the first and second semesters are core courses. In the third semester there will be three core theory courses and one elective theory course. College can choose any one of the elective courses given in Table 1. -

Organometallic Compounds
Chapter 11 Organometallic compounds Organometallics Reactions using organometallics Organometallic compounds Ch 11 #2 = comp’s containing a carbon-metal bond δ− δ+ C in organometallic comp’ds are nucleophilic. C in organic comp’d (like ROH, RNH , and RX) 2 δ+ δ are electrophilic. − due to ∆EN Ch 11 #3 in substitution reactions a carbon Nu: R-Li and R-MgX Ch 11 #4 (used to be) the two most common organometallics organolithium comp’ds BuLi = an alkyl lithium organomagnesium comp’ds = Grignard reagents 1912 Nobel Prize R, Ar, vinyl all possible; Br (as X) popular Ether (solvent) coordinates Mg, stabilizing it. Reactions of R-Li and R-MgX Ch 11 #5 reacts like a carbanion C:– ~ a C Nu: reactions as C Nu: (like SN(2)) nucleophilic addition to carbonyls ~ more often Chapt 16 Ch 11 #6 R-Li and R-MgX are very strong B: how strong? pKa? − − react even with very weak acid stronger than OH? NH2? useful for preparing deuterated HC Storage and reaction must be acid- and moisture-free. Transmetal(l)ation Ch 11 #7 R-Li is more reactive than R-MgX is. C–Li more polar than C–Mg C of R-Li more nu-philic [better Nu:] transmetalation [metal exchange] to less reactive [more stable] organometallic Coupling using Gilman reagent Ch 11 #8 coupling reaction (in organic chemistry) two hydrocarbon fragments are coupled (to form C–C) with the aid of a (transition) metal catalyst Gilman reagent coupling of R of R-X and R’ of Gilman reagent RX + R’2CuLi R–R’ mechanism? substitution of X with R’? not clear Ch 11 #9 R can be alkyl, aryl, or alkenyl [vinyl] which is not possible by R-Li or R-MgX Is R of Gilman why? they are SN(2). -
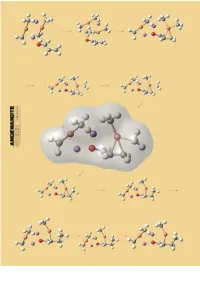
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

Studies Towards a Total Synthesis of Hippeastrine Johanna Myriam Helary 4769139
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of East Anglia digital repository Studies towards a total synthesis of Hippeastrine Johanna Myriam Helary 4769139 © I certify that the work contained in the thesis submitted by me for the degree of Master of Science by Research is my original work except where due reference is made to other authors, and has not been previously submitted by me for a degree at this or any other university. 1 Acknowledgements I would like to thanks my mum, my stepdad and Deeptee Horil Roy for their continued supports and encouragements over the last five years. For that I am very grateful. I would also like to thank all of the members Dr G. R Stephenson and Dr C. Richard with special thanks to Dr Julien Doulcet, Dr Paulina Glowacka and the rest of Floor 3 for their help, advices, patient explanations, and good humour over the last two years. Finally I would like to thank Dr. G. R Stephenson for all of his support, guidance and encouragements. In the loving memory of Sarah Delf, devoted friend and colleague. Wherever you are you always will be remembered. Rest in peace my amazing friend and fellow Hippeastrine crew member. 2 Table of Contents Abstract ................................................................................................................................. 5 1. Introduction .......................................................................................................................... 6 1.1. History of alkaloids ....................................................................................................... -

Hoto P QH PH
US010211400B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 ,211 , 400 B2 Hawker et al. ( 45 ) Date of Patent: Feb . 19 , 2019 (54 ) PHOTOPATTERNED GROWTH OF (51 ) Int . Ci. ELECTRONICALLY ACTIVE BRUSH HOIL 51/ 00 ( 2006 . 01 ) POLYMERS FOR LIGHT EMITTING DIODE C09K 11 / 06 (2006 . 01) DISPLAYS (Continued ) (71 ) Applicants : The Regents of the University of ( 52 ) U .S . CI. California , Oakland , CA (US ) ; Rohm CPC . .. .. HOIL 51 /0035 (2013 . 01 ) ; C09K 11/ 06 and Haas Electronic Materials LLC , ( 2013. 01 ) ; HOIL 51/ 0043 ( 2013 .01 ) ; Marlborough , MA (US ) ; Dow Global ( Continued ) Technologies LLC , Midland , MI (US ) (58 ) Field of Classification Search CPC .. .. .. HO1L 51 /0035 ; HO1L 51 /0043 ; HOIL ( 72 ) Inventors : Craig J . Hawker , Santa Barbara , CA (58 ) Fieldof c . 51. .HOZZ /5072 ; HOLLHOIL 551 / 5088 ; HO1L 51/ 5206 ; (US ) ; Zachariah Allen Page , Goleta , CA (US ) ; Peter Trefonas, III, Medway , (Continued ) MA (US ) ; Anatoliy N . Sokolov , Midland , MI (US ) ; John Kramer , ( 56 ) References Cited Midland , MI (US ) ; David S . Laitar, U . S . PATENT DOCUMENTS Midland , MI (US ) ; Sukrit Mukhopadhyay , Midland , MI (US ) ; 6 ,423 , 465 B1 7 /2002 Hawker et al . Benjaporn Narupai, Goleta , CA (US ) ; 6 , 447 , 879 B19 / 2002 Sakurai et al . Christian Wilhelm Pester , Goleta , CA ( Continued ) (US ) OTHER PUBLICATIONS (73 ) Assignees : DOW GLOBAL TECHNOLOGIES LLC , Midland , MI (US ) ; ROHM AND Choi et al. ; “ Simple Detachment Patterning of Organic Layers and HAAS ELECTRONIC MATERIALS Its Application to Organic Light- Emitting Diodes ” ; Adv. Mater. ; 17 , LLC , Marlborough , MA (US ) ; THE No . 2 ; Jan . 31 , 2005 , pp . 166 - 171 . REGENTS OF THE UNIVERSITY (Continued ) OF CALIFORNIA , Oakland, CA (US ) Primary Examiner — Su C Kim ( * ) Notice: Subject to any disclaimer, the term of this ( 74 ) Attorney , Agent , or Firm — Cantor Colburn LLP patent is extended or adjusted under 35 U . -

Organocuprate Aggregation and Reactivity: Decoding the 'Black Box'
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München Organocuprate Aggregation and Reactivity: Decoding the ‘Black Box’ Aliaksei Putau aus Vitebsk, Weißrussland 2012 Erklärung Diese Dissertation wurde im Sinne von § 7 der Promotionsordnung vom 28. November 2011 von Herrn Prof. Dr. Konrad Koszinowski betreut, und von Herrn Prof. Dr. Mayr von der Fakultät für Chemie und Pharmazie vertreten. Eidesstattliche Versicherung Diese Dissertation wurde selbständig, ohne unerlaubte Hilfe erarbeitet. München, 11.06.2012 _______________________ Aliaksei Putau Dissertation eingereicht am: 12.06.2012 1. Gutachter Prof. Dr. Herbert Mayr 2. Gutachter Prof. Dr. Konrad Koszinowski Mündliche Prüfung am: 20.07.2012 Acknowledgements It is a pleasure to thank the many people who made this project possible. My gratitude to my PhD supervisor, Prof. Dr. Konrad Koszinowski, is difficult to overstate. His sound advice, good company, and enthusiasm for trying new things ensured the tropical sunny climate in our group that is so necessary for fruitful (and, above all, enjoyable) research. Besides, his involvement in the group life outside the lab made all of us group members feel like team members, not just a loose bunch of colleagues. I am also deeply indebted to Prof. Dr. Herbert Mayr, not only for his continuous generous support and help, but also for running such a cool, all-star research group, which provided a great many social occasions. I thank SFB 749 for the financial support of this project, specifically Mrs. Birgit Carell, for her support and understanding of my worries and troubles of all kinds. Furthermore, my sincere gratitude goes to Prof. -

Select Reactions of Organoboranes and Organostannanes
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 8-2012 Select Reactions of Organoboranes and Organostannanes David W. Blevins [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Organic Chemistry Commons Recommended Citation Blevins, David W., "Select Reactions of Organoboranes and Organostannanes. " PhD diss., University of Tennessee, 2012. https://trace.tennessee.edu/utk_graddiss/1412 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by David W. Blevins entitled "Select Reactions of Organoboranes and Organostannanes." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Chemistry. George W. Kabalka, Major Professor We have read this dissertation and recommend its acceptance: Shane Foister, Craig Barnes, Kimberly Gwinn Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Select Reactions of Organoboranes and Organostannanes A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville David W. Blevins August 2012 DEDICATION I dedicate this work to Lisa, Preston, and Clarissa Blevins. ii ACKNOWLEDGEMENTS I would like to thank Dr. -

Decarboxylative Cross-Coupling: Bimetallic Nanoparticles As Catalysts and Low-Temperature Optimisation
Decarboxylative Cross-coupling: Bimetallic Nanoparticles as Catalysts and Low-Temperature Optimisation Joseph Kyle Sheppard Submitted in accordance with the requirements for the degree of Doctor of Philosophy University of Leeds School of Chemistry September 2020 The candidate confirms the work submitted is his own and that appropriate credit has been given where reference has been made to the work of others. I Acknowledgements First and foremost, I would like to thank my supervisors Dr. Bao N. Nguyen and Prof. Patrick McGowan for providing me with the opportunity to undertake this PhD. Bao, you deserve a medal for putting up with me for four whole years! Thank you dearly for everything you have taught me. Patrick, though our meetings were few and far between, I always left them brimming with new ideas and knowledge. I must also thank the wonderful Dr. Alexander Kulak; chiefly for all of your assistance with AAS and SEM analysis but also for being a friendly face that never failed to brighten my days. I’ll deeply miss the hours we spent, shouting over the noise of the spectrometer and trying to work out why it would develop an entirely new issue every time we used it. I would also like to thank Dr. Rebecca Stones for all of her hard work running my TEM samples and teaching me how to analyse the micrographs. Special thanks must also go to Dr. Mary Bayana for her patience in teaching me how to use all of the arcane and intricate analytical machines of the iPRD and to Martin Huscroft for helping me refine my HPLC method. -

Chem 341 • Organic Chemistry I Lecture Summary 29 • November 02, 2007
Chem 341 • Organic Chemistry I Lecture Summary 29 • November 02, 2007 Chapter 10 - Alkyl Halides Preparation of Organometallic Compounds - Grignard reagents Alkyl halides are good electrophiles. We will examine their reactivity in nucleophilic substitution in detail in the next chapter. Another useful property of alkyl halides as that reducing metals can insert between the C-X bond to prepare a C-metal bond. This drastically alters the reactivity as the metal is less electronegative than carbon. Thus, the reactivity of the carbon is reversed from being an electrophile to being a nucleophile. Grignard reagents are formed from the insertion of Mg metal into the C-X bond. These reagents are very reactive as bases and electrophiles and are extremely sensitive to any source of proton (water, ROH, RNH2, etc). This can be useful if one wants to reduce an alkyl halide to an alkane. It is also a useful method of introducing hydrogen isotopes, such as deuterium, by reaction of the Grignard reagent with heavy water (D stands for Deuterium, the isotope of hydrogen 2H). Grignard reagents can be prepared from a variety of different kinds of organohalides (eg. Alkyl halides, vinyl halides, etc.). One important reaction of Grignard reagents is their ability to add to carbonyl compounds (C=O double bonds). You will study this in detail next semester. δ− δ+ δ+ Br δ− Mg Br D Mg D2O O reactivity of OH carbon reversed R H then H+ R Preparation of Organometallic Compounds - Gilman reagents Lithium metal reacts with organohalides in a simlar fashion as magnesium. Organolithium reagents are very basic and are often used to deprotonated compounds that are not very acidic. -

REVIEW Methyl Complexes of the Transition Metals
REVIEW Methyl Complexes of the Transition Metals Jesús Campos,[b] Joaquín López-Serrano,[a] Riccardo Peloso,[a] and Ernesto Carmona*[a] Dedicated to Prof. Pierre Braunstein in recognition of his contributions to inorganic and organometallic chemistry [a] Title(s), Initial(s), Surname(s) of Author(s) including Corresponding Author(s) Department Institution Address 1 E-mail: [b] Title(s), Initial(s), Surname(s) of Author(s) Department Institution Address 2 Supporting information for this article is given via a link at the end of the document.((Please delete this text if not appropriate)) REVIEW Abstract: Organometallic chemistry can be considered as a wide focusing in the last 5 years. It begins with a brief description of area of knowledge that combines concepts of classic organic synthetic methods, followed by a selection of recently reported chemistry, i.e. based essentially on carbon, with molecular inorganic complexes with terminal and bridging methyl ligands from the chemistry, especially with coordination compounds. Transition metal groups 3 to 11 of the periodic table. A specific section is methyl complexes probably represent the simplest and most dedicated to methyl-bridged species with three-centre two- fundamental way to view how these two major areas of chemistry electron bonds. The review concludes highlighting relevant combine and merge into novel species with intriguing features in reactivity of the methyl group in this class of compounds. terms of reactivity, structure, and bonding. Citing more than 500 bibliographic references, this review aims to offer a concise view of recent advances in the field of transition metal complexes containing Ernesto Carmona (PhD degree, University M—CH3 fragments.