The Origins of Apicomplexan Sequence Innovation James Wasmuth1,*, Jennifer Daub1#, José Manuel Peregrín-Alvarez1,2, Constance A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Both LCCL-Domains of Human CRISPLD2 Have High Affinity for Lipid A
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimie 97 (2014) 66e71 Contents lists available at ScienceDirect Biochimie journal homepage: www.elsevier.com/locate/biochi Research paper Both LCCL-domains of human CRISPLD2 have high affinity for lipid A Viktor Vásárhelyi, Mária Trexler, László Patthy* Institute of Enzymology, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Budapest H-1518, PO Box 7, Hungary article info abstract Article history: The LCCL-domain is a recently defined protein module present in diverse extracellular multidomain Received 15 May 2013 proteins. Practically nothing is known about the molecular function of these domains; based on func- Accepted 20 September 2013 tional features of proteins harboring LCCL-domains it has been suggested that these domains might Available online 1 October 2013 function as lipopolysaccharide-binding domains. Here we show that the two LCCL-domains of human CRISPLD2 protein, a lipopolysaccharide-binding serum protein involved in defense against endotoxin Keywords: shock, have higher affinity for the lipid A, the toxic moiety of lipopolysaccharides than for ipopoly- CRISPLD2 protein saccharide. Our observation that the LCCL-domains of CRISPLD2 are specific for the toxic lipid A moiety of Endotoxin LCCL-domain the endotoxin suggests that it may block the interaction between endotoxins and the host endotoxin Lipid A receptors without interfering with the development of antibacterial immunity against the polysaccharide Lipopolysaccharide moiety of LPS. We suggest that the anti-inflammatory function of CRISPLD2 protein may account for its role in various pathological and developmental processes. Ó 2013 The Authors. -
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
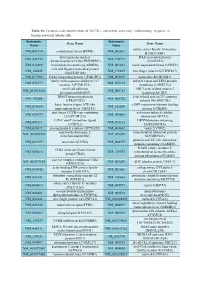
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

Analysis of the Plasmodium Falciparum Proteome by High-Accuracy Mass Organism
CORE Metadata, citation and similar papers at core.ac.uk Provided by University of Southern Denmark Research Output letters to nature 12. Lasonder, E. et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass organism. Here we describe a large-scale, high-accuracy (average spectrometry. Nature 419, 537–542 (2002). 13. Ewing, B. & Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. deviation less than 0.02 Da at 1,000 Da) mass spectrometric 1–3 Genome Res. 8, 186–194 (1998). proteome analysis of selected stages of the human malaria 14. Oefner, P. J. et al. Efficient random subcloning of DNA sheared in a recirculating point-sink flow parasite Plasmodium falciparum. The analysis revealed 1,289 system. Nucleic Acids Res. 24, 3879–3886 (1996). proteins of which 714 proteins were identified in asexual blood 15. Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998). stages, 931 in gametocytes and 645 in gametes. The last two 16. Gordon, D., Abajian, C. & Green, P. Consed: a graphical tool for sequence finishing. Genome Res. 8, groups provide insights into the biology of the sexual stages of 195–202 (1998). the parasite, and include conserved, stage-specific, secreted and 17. Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. membrane-associated proteins. A subset of these proteins con- J. Mol. Biol. 215, 403–410 (1990). 18. Apweiler, R. et al. The InterPro database, an integrated documentation resource for protein families, tain domains that indicate a role in cell–cell interactions, and domains and functional sites. -

Cleaved Cochlin Sequesters Pseudomonas Aeruginosa and Activates Innate Immunity in the Inner Ear
Article Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear Graphical Abstract Authors Jinsei Jung, Jee Eun Yoo, Young Ho Choe, ..., Joo-Heon Yoon, Young-Min Hyun, Jae Young Choi Correspondence [email protected] (Y.-M.H.), [email protected] (J.Y.C.) In Brief Jung et al. show that the cleaved cochlin LCCL domain enhances innate immune responses in the inner ear by aggregating infiltrated bacteria and recruiting innate immune cells. This spatiotemporally protective function of LCCL protects hearing function in the organ of Corti against effects of bacterial invasion in the inner ear. Highlights d Cleaved inner ear cochlin LCCL secretes to perilymph space post-bacterial infection d LCCL induces bacterial aggregation in the scala tympani d Spatiotemporal innate immune response by LCCL protects the sensory organ of Corti À À d LCCL rescues Coch / mouse post-Pseudomonas inner ear infection hearing loss Jung et al., 2019, Cell Host & Microbe 25, 1–13 April 10, 2019 ª 2019 Elsevier Inc. https://doi.org/10.1016/j.chom.2019.02.001 Please cite this article in press as: Jung et al., Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear, Cell Host & Microbe (2019), https://doi.org/10.1016/j.chom.2019.02.001 Cell Host & Microbe Article Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear Jinsei Jung,1,2,7 Jee Eun Yoo,1,2,7 Young Ho Choe,3,4 Sang Chul Park,1 Hyun Jae Lee,1,2 Hack June Lee,1,2 Byunghwa Noh,1,2 Sung Huhn Kim,1,2 -

Subcellular Localisation, Secretion, and Post-Translational Processing Of
479 ORIGINAL ARTICLE J Med Genet: first published as 10.1136/jmg.40.7.479 on 1 July 2003. Downloaded from Subcellular localisation, secretion, and post-translational processing of normal cochlin, and of mutants causing the sensorineural deafness and vestibular disorder, DFNA9 N G Robertson, S A Hamaker, V Patriub, J C Aster, C C Morton ............................................................................................................................. J Med Genet 2003;40:479–486 Five missense mutations in the FCH/LCCL domain of the COCH gene, encoding the protein cochlin, are pathogenic for the autosomal dominant hearing loss and vestibular dysfunction disorder, DFNA9. To date, the function of cochlin and the mechanism of pathogenesis of the mutations are unknown. We have used the biological system of transient transfections of the entire protein coding region of COCH into several mammalian cell lines, to investigate various functional properties of cochlin. By western blot analysis of lysates prepared from transfected cells, we show that cochlin is a secreted protein. Immuno- cytochemistry shows concentrated localisation of cochlin in perinuclear structures consistent with the Golgi apparatus and endoplasmic reticulum, showing intracellular passage through these secretory compartments. We detected that cochlin is proteolytically cleaved between the FCH/LCCL domain and the downstream vWFA domains, resulting in a smaller cochlin isoform of ∼50 kDa. Interestingly, this isoform lacks the entire mutation bearing FCH/LCCL domain. We have also shown that cochlin is See end of article for N-glycosylated in its mature secreted form. Previous studies of the FCH/LCCL domain alone, expressed authors’ affiliations in bacteria, have demonstrated that three of four DFNA9 mutations cause misfolding of this domain. -

Does Otovestibular Loss in the Autosomal Dominant Disorder DFNA9 Have an Impact of on Cognition? a Systematic Review
SYSTEMATIC REVIEW published: 09 January 2018 doi: 10.3389/fnins.2017.00735 Does Otovestibular Loss in the Autosomal Dominant Disorder DFNA9 Have an Impact of on Cognition? A Systematic Review Jonas De Belder 1†, Stijn Matthysen 1†, Annes J. Claes 1, 2, Griet Mertens 1, 2, Paul Van de Heyning 1, 2 and Vincent Van Rompaey 1, 2* 1 Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium, 2 Department of Otorhinolaryngology and Head and Neck Surgery, Antwerp University Hospital, Edegem, Belgium Edited by: Background and Purpose: Cognitive impairment has been observed in patients with Etienne De Villers-Sidani, bilateral vestibular loss (BVL) and in patients with sensorineural hearing loss (SNHL). McGill University, Canada DFNA9 is an autosomal dominant disorder that causes a combination of both sensory Reviewed by: deficits by the 3rd to 5th decade. We therefore hypothesize a combined detrimental effect Martha M. Shiell, Maastricht University, Netherlands on cognition. The aim of this systematic review was to identify studies related to DFNA9 Ronald Pennings, in general and its relationship with cognitive impairment more specifically. Radboud University Nijmegen Medical Center, Netherlands Materials and Methods: Several databases including Medline, Cochrane Database *Correspondence: of Systematic Reviews, Cochrane Central Register of Controlled Trials, ISI Web of Vincent Van Rompaey Knowledge, and Web of Science were searched to accumulate information about [email protected] DFNA9-mutations, including phenotype, genotype, pathophysiology, quality of life (QOL), †These authors have contributed equally to this work. and imaging in general and cognitive function more specifically. A qualitative analysis was performed on the 55 articles that qualified. -

Protein to Regulate Endotoxin Function Protein CRISPLD2 Is A
The Novel Lipopolysaccharide-Binding Protein CRISPLD2 Is a Critical Serum Protein to Regulate Endotoxin Function This information is current as Zhi-Qin Wang, Wen-Ming Xing, Hua-Hua Fan, Ke-Sheng of September 29, 2021. Wang, Hai-Kuo Zhang, Qin-Wan Wang, Jia Qi, Hong-Meng Yang, Jie Yang, Ya-Na Ren, Shu-Jian Cui, Xin Zhang, Feng Liu, Dao-Hong Lin, Wen-Hui Wang, Michael K. Hoffmann and Ze-Guang Han J Immunol 2009; 183:6646-6656; Prepublished online 28 Downloaded from October 2009; doi: 10.4049/jimmunol.0802348 http://www.jimmunol.org/content/183/10/6646 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2009/10/28/jimmunol.080234 Material 8.DC1 References This article cites 46 articles, 12 of which you can access for free at: http://www.jimmunol.org/content/183/10/6646.full#ref-list-1 Why The JI? Submit online. by guest on September 29, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. -

Domain Shuffling and the Evolution of Vertebrates
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Domain shuffling and the evolution of vertebrates Takeshi Kawashima1, 2, 3, Shuichi Kawashima4, Chisaki Tanaka5, Miho Murai6, Masahiko Yoneda6, Nicholas H. Putnam2, Daniel S. Rokhsar2, Minoru Kanehisa4,7, Nori Satoh1, 8, Hiroshi Wada5 1: Okinawa Institute of Science and Technology, Uruma, Okinawa, 904-2234, Japan 2: Department of Energy Joint Genome Institute, Walnut Creek, CA 94598, USA 3: Japanese Society for Promotion of sciences, 6 Ichibancho, Chiyoda-ku, Tokyo, 102-8471, Japan 4: Human Genome Center, Institute of Medical Science, University of Tokyo, 4-6-1 Shirokane-dai Minato-ku Tokyo 108-8639, Japan 5: Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba 305-8572 JAPAN 6: Aichi Prefectural College of Nursing and Health, Miriyama-ku Nagoya 463-8502 JAPAN 7: Bioinformatics Center, Institute of Chemical Research, Kyoto University, Kyoto 611-0011, Japan 8: Department of Zoology, Graduate School of Sciences, Kyoto University, Kyoto 606-8502, JAPAN Address for correspondense: Hiroshi Wada Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba 305-8572 JAPAN E-mail: [email protected], Tel & Fax: +81-29-853-4671 Takeshi Kawashima Okinawa Institute of Science and Technology, Uruma, Okinawa, 904-2234, Japan E-mail: [email protected], Tel; +1-98-921-2245, Fax; +1-98-934-5622 Running title: Domain shuffling in vertebrate genomes Key words: domain shuffling, exon shuffling, vertebrate, cartilage, chordate, notochord, ascidian, amphioxus 1 Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The evolution of vertebrates included a number of important events: the development of cartilage, the immune system, and complicated craniofacial structures. -

Integrated Bioinformatics Analysis Reveals Novel Key Biomarkers and Potential Candidate Small Molecule Drugs in Gestational Diabetes Mellitus
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434569; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Integrated bioinformatics analysis reveals novel key biomarkers and potential candidate small molecule drugs in gestational diabetes mellitus Basavaraj Vastrad1, Chanabasayya Vastrad*2, Anandkumar Tengli3 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. 3. Department of Pharmaceutical Chemistry, JSS College of Pharmacy, Mysuru and JSS Academy of Higher Education & Research, Mysuru, Karnataka, 570015, India * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434569; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Gestational diabetes mellitus (GDM) is one of the metabolic diseases during pregnancy. The identification of the central molecular mechanisms liable for the disease pathogenesis might lead to the advancement of new therapeutic options. The current investigation aimed to identify central differentially expressed genes (DEGs) in GDM. The transcription profiling by array data (E-MTAB-6418) was obtained from the ArrayExpress database. The DEGs between GDM samples and non GDM samples were analyzed with limma package. Gene ontology (GO) and REACTOME enrichment analysis were performed using ToppGene. Then we constructed the protein-protein interaction (PPI) network of DEGs by the Search Tool for the Retrieval of Interacting Genes database (STRING) and module analysis was performed. -

A Novel COCH Mutation, V104del, Impairs Folding of the LCCL Domain
1of3 J Med Genet: first published as 10.1136/jmg.2003.012286 on 16 January 2004. Downloaded from ONLINE MUTATION REPORT A novel COCH mutation, V104del, impairs folding of the LCCL domain of cochlin and causes progressive hearing loss I Nagy, M Horva´th, M Trexler, G Re´pa´ssy, L Patthy ............................................................................................................................... J Med Genet 2004;41:e9 (http://www.jmedgenet.com/cgi/content/full/41/1/e9) ecently the causative gene of autosomal dominant sensorineural nonsyndromic late onset hearing loss Key points R(DFNA9) has been identified as the COCH gene, which lies in the DFNA9 region of human chromosome 14 (gene N Autosomal dominant nonsyndromic sensorineural map locus 14q12–q13). Molecular analysis of cases of DFNA9 deafness 9 (DFNA9, OMIM #601369) is a rare, late have identified several families with five different mutations onset hearing loss. In DFNA9 patients, hearing loss in this gene.1–5 usually begins in the third decade of life as a high The cochlin protein encoded by COCH is an extracellular frequency loss with concomitant vestibular dysfunction. matrix protein that contains an LCCL domain and two von N 1 DFNA9 is caused by mutations in the COCH gene, Willebrand type A domains. Interestingly, all mutations which encodes cochlin, an extracellular matrix protein causing DFNA9 type deafness disorder affect the LCCL that contains an LCCL domain and two von Willebrand domain of cochlin. In view of the central role of this domain type A domains. in the DFNA9 disorder, we initiated studies on it to explore the molecular basis of the disease.6 We found that most N Molecular analysis of cases of DFNA9 has identified DFNA9 mutations affected conserved structural elements of five different mutations in this gene. -

Dcbld2/Esdn Is Essential for Proper Optic Tract Formation and Retinal Lamination Ryan Mears Joy University of Vermont
University of Vermont ScholarWorks @ UVM Graduate College Dissertations and Theses Dissertations and Theses 2016 Dcbld2/esdn Is Essential For Proper Optic Tract Formation And Retinal Lamination Ryan Mears Joy University of Vermont Follow this and additional works at: https://scholarworks.uvm.edu/graddis Part of the Cell and Developmental Biology Commons Recommended Citation Joy, Ryan Mears, "Dcbld2/esdn Is Essential For Proper Optic Tract Formation And Retinal Lamination" (2016). Graduate College Dissertations and Theses. 563. https://scholarworks.uvm.edu/graddis/563 This Thesis is brought to you for free and open access by the Dissertations and Theses at ScholarWorks @ UVM. It has been accepted for inclusion in Graduate College Dissertations and Theses by an authorized administrator of ScholarWorks @ UVM. For more information, please contact [email protected]. DCBLD2/ESDN IS ESSENTIAL FOR PROPER OPTIC TRACT FORMATION AND RETINAL LAMINATION A Thesis Presented by Ryan Joy to The Faculty of the Graduate College of The University of Vermont In Partial Fulfillment of the Requirements for the Degree of Master of Science Specializing in Biology May, 2016 Defense Date: January 7, 2016 Thesis Examination Committee: Bryan A. Ballif, Ph.D., Advisor Jason Stumpff, Ph.D., Chairperson Alicia M. Ebert, Ph.D. Paula Deming, Ph.D., MT Cynthia J. Forehand, Ph.D., Dean of the Graduate College ABSTRACT The Discoidin, CUB and LCCL domain-containing protein 2 (DCBLD2/ESDN/CLCP1) is a type-I, transmembrane receptor that mediates diverse cellular processes such as angiogenesis, vascular remodeling, cellular migration and proliferation. Identification of DCBLD2 in a proteomics screen to identify substrates of Src family tyrosine kinases that bind the Src homology 2 domain of CT10 regulator of kinase-Like (CrkL), a critical scaffolding protein for neuronal development, led to a need for further characterization of the protein. -
Overview of the Matrisome—An Inventory of Extracellular Matrix Constituents and Functions
Downloaded from http://cshperspectives.cshlp.org/ on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press Overview of the Matrisome—An Inventory of Extracellular Matrix Constituents and Functions Richard O. Hynes and Alexandra Naba Howard Hughes Medical Institute, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 Correspondence: [email protected] Completion of genome sequences for many organisms allows a reasonably complete definition of the complement of extracellular matrix (ECM) proteins. In mammals this “core matrisome” comprises 300 proteins. In addition there are large numbers of ECM- modifying enzymes, ECM-binding growth factors, and other ECM-associated proteins. These different categories of ECM and ECM-associated proteins cooperate to assemble and remodel extracellular matrices and bind to cells through ECM receptors. Togetherwith recep- tors for ECM-bound growth factors, they provide multiple inputs into cells to control survival, proliferation, differentiation, shape, polarity, and motility of cells. The evolution of ECM pro- teins was key in the transition to multicellularity, the arrangement of cells into tissue layers, and the elaboration of novel structures during vertebrate evolution. This key role of ECM is reflected in the diversity of ECM proteins and the modular domain structures of ECM proteins both allow their multiple interactions and, during evolution, development of novel protein architectures. he term extracellular matrix (ECM) means make up basement membranes. Biochemistry Tsomewhat different things to different of native ECM was, and still is, impeded by people (Hay 1981, 1991; Mecham 2011). Light the fact that the ECM is, by its very nature, and electron microscopy show that extracellular insoluble and is frequently cross-linked.