A Novel COCH Mutation, V104del, Impairs Folding of the LCCL Domain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cochlin in the Eye: Functional Implications
Cochlin in the eye: Functional implications Picciani, R., Desaia, K., Guduric-Fuchs, J., Cogliati, T., Morton, C. C., & Bhattacharya, S. K. (2007). Cochlin in the eye: Functional implications. Progress in Retinal and Eye Research, 26 (5)(5), 453-469. https://doi.org/10.1016/j.preteyeres.2007.06.002 Published in: Progress in Retinal and Eye Research Queen's University Belfast - Research Portal: Link to publication record in Queen's University Belfast Research Portal General rights Copyright for the publications made accessible via the Queen's University Belfast Research Portal is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The Research Portal is Queen's institutional repository that provides access to Queen's research output. Every effort has been made to ensure that content in the Research Portal does not infringe any person's rights, or applicable UK laws. If you discover content in the Research Portal that you believe breaches copyright or violates any law, please contact [email protected]. Download date:26. Sep. 2021 Author’s Accepted Manuscript Cochlin in the eye: Functional implications Renata Picciani, Kavita Desai, Jasenka Guduric- Fuchs,Tiziana Cogliati, Cynthia C. Morton, Sanjoy K. Bhattacharya PII: S1350-9462(07)00040-7 DOI: doi:10.1016/j.preteyeres.2007.06.002 Reference: JPRR 345 www.elsevier.com/locate/prer To appear in: Progress in Retinal and Eye Research Cite this article as: Renata Picciani, Kavita Desai, Jasenka Guduric-Fuchs, Tiziana Cogliati, Cynthia C. -

Both LCCL-Domains of Human CRISPLD2 Have High Affinity for Lipid A
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimie 97 (2014) 66e71 Contents lists available at ScienceDirect Biochimie journal homepage: www.elsevier.com/locate/biochi Research paper Both LCCL-domains of human CRISPLD2 have high affinity for lipid A Viktor Vásárhelyi, Mária Trexler, László Patthy* Institute of Enzymology, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Budapest H-1518, PO Box 7, Hungary article info abstract Article history: The LCCL-domain is a recently defined protein module present in diverse extracellular multidomain Received 15 May 2013 proteins. Practically nothing is known about the molecular function of these domains; based on func- Accepted 20 September 2013 tional features of proteins harboring LCCL-domains it has been suggested that these domains might Available online 1 October 2013 function as lipopolysaccharide-binding domains. Here we show that the two LCCL-domains of human CRISPLD2 protein, a lipopolysaccharide-binding serum protein involved in defense against endotoxin Keywords: shock, have higher affinity for the lipid A, the toxic moiety of lipopolysaccharides than for ipopoly- CRISPLD2 protein saccharide. Our observation that the LCCL-domains of CRISPLD2 are specific for the toxic lipid A moiety of Endotoxin LCCL-domain the endotoxin suggests that it may block the interaction between endotoxins and the host endotoxin Lipid A receptors without interfering with the development of antibacterial immunity against the polysaccharide Lipopolysaccharide moiety of LPS. We suggest that the anti-inflammatory function of CRISPLD2 protein may account for its role in various pathological and developmental processes. Ó 2013 The Authors. -
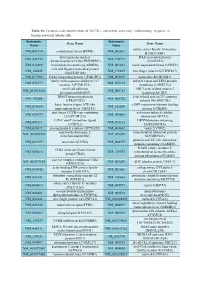
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

Analysis of the Plasmodium Falciparum Proteome by High-Accuracy Mass Organism
CORE Metadata, citation and similar papers at core.ac.uk Provided by University of Southern Denmark Research Output letters to nature 12. Lasonder, E. et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass organism. Here we describe a large-scale, high-accuracy (average spectrometry. Nature 419, 537–542 (2002). 13. Ewing, B. & Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. deviation less than 0.02 Da at 1,000 Da) mass spectrometric 1–3 Genome Res. 8, 186–194 (1998). proteome analysis of selected stages of the human malaria 14. Oefner, P. J. et al. Efficient random subcloning of DNA sheared in a recirculating point-sink flow parasite Plasmodium falciparum. The analysis revealed 1,289 system. Nucleic Acids Res. 24, 3879–3886 (1996). proteins of which 714 proteins were identified in asexual blood 15. Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998). stages, 931 in gametocytes and 645 in gametes. The last two 16. Gordon, D., Abajian, C. & Green, P. Consed: a graphical tool for sequence finishing. Genome Res. 8, groups provide insights into the biology of the sexual stages of 195–202 (1998). the parasite, and include conserved, stage-specific, secreted and 17. Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. membrane-associated proteins. A subset of these proteins con- J. Mol. Biol. 215, 403–410 (1990). 18. Apweiler, R. et al. The InterPro database, an integrated documentation resource for protein families, tain domains that indicate a role in cell–cell interactions, and domains and functional sites. -

Sequence Function Classification by Machine Learning Methods
From the Institute for Neuro- and Bioinformatics of the University of Lübeck Director: Prof. Dr. Thomas Martinetz Sequence function classification by machine learning methods Dissertation for Fulfillment of Requirements for the Doctoral Degree of the University of Lübeck from the Department of Computer Sciences/Engineering Submitted by Krishna Kumar Kandaswamy Born in Coimbatore - TN, India Lübeck, 2011 First referee: Prof. Thomas Martinetz Second referee: Prof. Enno Hartmann Date of oral examination: 18.04.2012 Approved for printing, Lübeck : 23.04.2012 Acknowledgements Many people contributed either directly or indirectly. It will be difficult to name all of them. To start, I express sincere gratitude to my thesis advisor Prof. Thomas Martinetz, I enjoyed every moment discussing many related things to my thesis. I always admired his perfect scientific judgment in solving problems, a clear wit and a rare ability to stay cool in the most critical situations. Despite his busy schedule, he was always there to advise and give support whenever required. I convey my thanks and gratitude to Prof. Enno Hartmann, who introduced me to the field of protein secretion and translocation. In a budding phase, we had many discussions and I used to wonder his ability to put in tireless efforts in reading and discussing things out (which helped in speeding up the things). I am very grateful to Dr. Kai-Uwe Kalies and Dr. Steffen Möller for an incalculable number of suggestions, critical talk and motivational sessions. A special thank you must go to Dr. Ganesan Pugalenthi for his valuable inputs in regards to the concept of sequence analysis. -

(12) Patent Application Publication (10) Pub. No.: US 2003/0198970 A1 Roberts (43) Pub
US 2003O19897OA1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0198970 A1 Roberts (43) Pub. Date: Oct. 23, 2003 (54) GENOSTICS clinical trials on groups or cohorts of patients. This group data is used to derive a Standardised method of treatment (75) Inventor: Gareth Wyn Roberts, Cambs (GB) which is Subsequently applied on an individual basis. There is considerable evidence that a significant factor underlying Correspondence Address: the individual variability in response to disease, therapy and FINNEGAN, HENDERSON, FARABOW, prognosis lies in a person's genetic make-up. There have GARRETT & DUNNER been numerous examples relating that polymorphisms LLP within a given gene can alter the functionality of the protein 1300 ISTREET, NW encoded by that gene thus leading to a variable physiological WASHINGTON, DC 20005 (US) response. In order to bring about the integration of genomics into medical practice and enable design and building of a (73) Assignee: GENOSTIC PHARMA LIMITED technology platform which will enable the everyday practice (21) Appl. No.: 10/206,568 of molecular medicine a way must be invented for the DNA Sequence data to be aligned with the identification of genes (22) Filed: Jul. 29, 2002 central to the induction, development, progression and out come of disease or physiological States of interest. Accord Related U.S. Application Data ing to the invention, the number of genes and their configu rations (mutations and polymorphisms) needed to be (63) Continuation of application No. 09/325,123, filed on identified in order to provide critical clinical information Jun. 3, 1999, now abandoned. concerning individual prognosis is considerably less than the 100,000 thought to comprise the human genome. -

Uniprot Acceprotiens 121 113 Ratio(113/12 114 Ratio
Uniprot Acceprotiens 121 113 ratio(113/12 114 ratio(114/12 115 ratio(115/12 116 ratio(116/12 117 ratio(117/12 118 ratio(118/12 119 ratio(119/121) P02768 Serum albumin OS=Homo s666397.2 862466.6 1.29 593482.1 0.89 2220420.5 3.33 846469.3 1.27 634302.5 0.95 736961.1 1.11 842297.5 1.26 P02760 Protein AMBP OS=Homo s381627.7 294812.3 0.77 474165.8 1.24 203377.3 0.53 349197.6 0.92 346271.7 0.91 328356.1 0.86 411229.3 1.08 B4E1B2 cDNA FLJ53691, highly sim78511.8 107560.1 1.37 85218.8 1.09 199640.4 2.54 90022.3 1.15 73427.3 0.94 82722 1.05 102491.8 1.31 A0A0K0K1HEpididymis secretory sperm 3358.1 4584.8 1.37 4234.8 1.26 8496.1 2.53 4193.7 1.25 3507.1 1.04 3632.2 1.08 4873.3 1.45 D3DNU8 Kininogen 1, isoform CRA_302648.3 294936.6 0.97 257956.9 0.85 193831.3 0.64 290406.7 0.96 313453.3 1.04 279805.5 0.92 228883.9 0.76 B4E1C2 Kininogen 1, isoform CRA_167.9 229.7 1.37 263.2 1.57 278 1.66 326 1.94 265.5 1.58 290.3 1.73 341.5 2.03 O60494 Cubilin OS=Homo sapiens G40132.6 45037.5 1.12 38654.5 0.96 34055.8 0.85 39708.6 0.99 44702.9 1.11 45025.7 1.12 32701.3 0.81 P98164 Low-density lipoprotein rece40915.4 45344.8 1.11 35817.7 0.88 35721.8 0.87 42157.7 1.03 46693.4 1.14 48624 1.19 38847.7 0.95 A0A024RABHeparan sulfate proteoglyca46985.3 43536.1 0.93 49827.7 1.06 33964.3 0.72 44780.9 0.95 46858.6 1.00 47703.5 1.02 37785.7 0.80 P01133 Pro-epidermal growth factor 75270.8 73109.5 0.97 66336.1 0.88 56680.9 0.75 70877.8 0.94 76444.3 1.02 81110.3 1.08 65749.7 0.87 Q6N093 Putative uncharacterized pro47825.3 55632.5 1.16 48428.3 1.01 63601.5 1.33 65204.2 1.36 59384.5 -

Molecular Architecture Underlying Fluid Absorption by the Developing Inner
RESEARCH ARTICLE Molecular architecture underlying fluid absorption by the developing inner ear Keiji Honda1†, Sung Huhn Kim2‡, Michael C Kelly3, Joseph C Burns3§, Laura Constance2, Xiangming Li2#, Fei Zhou2, Michael Hoa4, Matthew W Kelley3, Philine Wangemann2*, Robert J Morell5, Andrew J Griffith1* 1Molecular Biology and Genetics Section, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, United States; 2Anatomy and Physiology Department, Kansas State University, Manhattan, United States; 3Developmental Neuroscience Section, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, United States; 4Auditory Development and Restoration Program, National Institute on *For correspondence: Deafness and Other Communication Disorders, National Institutes of Health, [email protected] (PW); Bethesda, United States; 5Genomics and Computational Biology Core, National [email protected] (AJG) Institute on Deafness and Other Communication Disorders, National Institutes of Present address: Health, Bethesda, United States †Otolaryngology Department, Tsuchiura Kyodo General Hospital, Tsuchiura, Japan; ‡Department of Abstract Mutations of SLC26A4 are a common cause of hearing loss associated with Otorhinolaryngology, Head and enlargement of the endolymphatic sac (EES). Slc26a4 expression in the developing mouse Neck Surgery, Yonsei University College of Medicine, Seoul, endolymphatic sac is required for acquisition of normal inner ear structure and function. Here, we Korea; §Decibel Therapeutics, show that the mouse endolymphatic sac absorbs fluid in an SLC26A4-dependent fashion. Fluid Cambridge, United States; absorption was sensitive to ouabain and gadolinium but insensitive to benzamil, bafilomycin and #Technique R and D-Drug S3226. Single-cell RNA-seq analysis of pre- and postnatal endolymphatic sacs demonstrates two Substance, GlaxoSmithKline types of differentiated cells. -

Investigating the Effect of Chronic Activation of AMP-Activated Protein
Investigating the effect of chronic activation of AMP-activated protein kinase in vivo Alice Pollard CASE Studentship Award A thesis submitted to Imperial College London for the degree of Doctor of Philosophy September 2017 Cellular Stress Group Medical Research Council London Institute of Medical Sciences Imperial College London 1 Declaration I declare that the work presented in this thesis is my own, and that where information has been derived from the published or unpublished work of others it has been acknowledged in the text and in the list of references. This work has not been submitted to any other university or institute of tertiary education in any form. Alice Pollard The copyright of this thesis rests with the author and is made available under a Creative Commons Attribution Non-Commercial No Derivatives license. Researchers are free to copy, distribute or transmit the thesis on the condition that they attribute it, that they do not use it for commercial purposes and that they do not alter, transform or build upon it. For any reuse or redistribution, researchers must make clear to others the license terms of this work. 2 Abstract The prevalence of obesity and associated diseases has increased significantly in the last decade, and is now a major public health concern. It is a significant risk factor for many diseases, including cardiovascular disease (CVD) and type 2 diabetes. Characterised by excess lipid accumulation in the white adipose tissue, which drives many associated pathologies, obesity is caused by chronic, whole-organism energy imbalance; when caloric intake exceeds energy expenditure. Whilst lifestyle changes remain the most effective treatment for obesity and the associated metabolic syndrome, incidence continues to rise, particularly amongst children, placing significant strain on healthcare systems, as well as financial burden. -

Cleaved Cochlin Sequesters Pseudomonas Aeruginosa and Activates Innate Immunity in the Inner Ear
Article Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear Graphical Abstract Authors Jinsei Jung, Jee Eun Yoo, Young Ho Choe, ..., Joo-Heon Yoon, Young-Min Hyun, Jae Young Choi Correspondence [email protected] (Y.-M.H.), [email protected] (J.Y.C.) In Brief Jung et al. show that the cleaved cochlin LCCL domain enhances innate immune responses in the inner ear by aggregating infiltrated bacteria and recruiting innate immune cells. This spatiotemporally protective function of LCCL protects hearing function in the organ of Corti against effects of bacterial invasion in the inner ear. Highlights d Cleaved inner ear cochlin LCCL secretes to perilymph space post-bacterial infection d LCCL induces bacterial aggregation in the scala tympani d Spatiotemporal innate immune response by LCCL protects the sensory organ of Corti À À d LCCL rescues Coch / mouse post-Pseudomonas inner ear infection hearing loss Jung et al., 2019, Cell Host & Microbe 25, 1–13 April 10, 2019 ª 2019 Elsevier Inc. https://doi.org/10.1016/j.chom.2019.02.001 Please cite this article in press as: Jung et al., Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear, Cell Host & Microbe (2019), https://doi.org/10.1016/j.chom.2019.02.001 Cell Host & Microbe Article Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear Jinsei Jung,1,2,7 Jee Eun Yoo,1,2,7 Young Ho Choe,3,4 Sang Chul Park,1 Hyun Jae Lee,1,2 Hack June Lee,1,2 Byunghwa Noh,1,2 Sung Huhn Kim,1,2 -

Subcellular Localisation, Secretion, and Post-Translational Processing Of
479 ORIGINAL ARTICLE J Med Genet: first published as 10.1136/jmg.40.7.479 on 1 July 2003. Downloaded from Subcellular localisation, secretion, and post-translational processing of normal cochlin, and of mutants causing the sensorineural deafness and vestibular disorder, DFNA9 N G Robertson, S A Hamaker, V Patriub, J C Aster, C C Morton ............................................................................................................................. J Med Genet 2003;40:479–486 Five missense mutations in the FCH/LCCL domain of the COCH gene, encoding the protein cochlin, are pathogenic for the autosomal dominant hearing loss and vestibular dysfunction disorder, DFNA9. To date, the function of cochlin and the mechanism of pathogenesis of the mutations are unknown. We have used the biological system of transient transfections of the entire protein coding region of COCH into several mammalian cell lines, to investigate various functional properties of cochlin. By western blot analysis of lysates prepared from transfected cells, we show that cochlin is a secreted protein. Immuno- cytochemistry shows concentrated localisation of cochlin in perinuclear structures consistent with the Golgi apparatus and endoplasmic reticulum, showing intracellular passage through these secretory compartments. We detected that cochlin is proteolytically cleaved between the FCH/LCCL domain and the downstream vWFA domains, resulting in a smaller cochlin isoform of ∼50 kDa. Interestingly, this isoform lacks the entire mutation bearing FCH/LCCL domain. We have also shown that cochlin is See end of article for N-glycosylated in its mature secreted form. Previous studies of the FCH/LCCL domain alone, expressed authors’ affiliations in bacteria, have demonstrated that three of four DFNA9 mutations cause misfolding of this domain. -

Does Otovestibular Loss in the Autosomal Dominant Disorder DFNA9 Have an Impact of on Cognition? a Systematic Review
SYSTEMATIC REVIEW published: 09 January 2018 doi: 10.3389/fnins.2017.00735 Does Otovestibular Loss in the Autosomal Dominant Disorder DFNA9 Have an Impact of on Cognition? A Systematic Review Jonas De Belder 1†, Stijn Matthysen 1†, Annes J. Claes 1, 2, Griet Mertens 1, 2, Paul Van de Heyning 1, 2 and Vincent Van Rompaey 1, 2* 1 Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium, 2 Department of Otorhinolaryngology and Head and Neck Surgery, Antwerp University Hospital, Edegem, Belgium Edited by: Background and Purpose: Cognitive impairment has been observed in patients with Etienne De Villers-Sidani, bilateral vestibular loss (BVL) and in patients with sensorineural hearing loss (SNHL). McGill University, Canada DFNA9 is an autosomal dominant disorder that causes a combination of both sensory Reviewed by: deficits by the 3rd to 5th decade. We therefore hypothesize a combined detrimental effect Martha M. Shiell, Maastricht University, Netherlands on cognition. The aim of this systematic review was to identify studies related to DFNA9 Ronald Pennings, in general and its relationship with cognitive impairment more specifically. Radboud University Nijmegen Medical Center, Netherlands Materials and Methods: Several databases including Medline, Cochrane Database *Correspondence: of Systematic Reviews, Cochrane Central Register of Controlled Trials, ISI Web of Vincent Van Rompaey Knowledge, and Web of Science were searched to accumulate information about [email protected] DFNA9-mutations, including phenotype, genotype, pathophysiology, quality of life (QOL), †These authors have contributed equally to this work. and imaging in general and cognitive function more specifically. A qualitative analysis was performed on the 55 articles that qualified.