Gene Diversity Explains Variation in Biological Features of Insect Killing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Use of Entomopathogenic Fungi for Fruit Fly Control in Area-Wide SIT Programmes
Use of Entomopathogenic Fungi for Fruit Fly Control in Area-Wide SIT Programmes Food and Agriculture Organization of the United Nations International Atomic Energy Agency Vienna, 2019 DISCLAIMER The material in this document has been supplied by the authors. The views expressed remain the responsibility of the authors and do not necessarily reflect those of the government(s) or the designating Member State(s). In particular, the FAO, IAEA, nor any other organization or body sponsoring the development of this document can be held responsible for any material reproduced in the document. The proper citation for this document is: FAO/IAEA. 2019. Use of Entomopathogenic Fungi for Fruit Fly Control in Area-Wide SIT Programmes, A. Villaseñor, S. Flores, S. E. Campos, J. Toledo, P. Montoya, P. Liedo and W. Enkerlin (eds.), Food and Agriculture Organization of the United Nations/International Atomic Energy Agency. Vienna, Austria. 43 pp. Use of Entomopathogenic Fungi for Fruit Fly Control in Area- wide SIT Programmes Antonio Villaseñor IAEA/IPCS Consultant San Pedro La Laguna, Nayarit, México Salvador Flores Programa Moscafrut SADER-SENASICA, Metapa, Chiapas, México Sergio E. Campos Programa Moscafrut SADER-SENASICA, Metapa, Chiapas, México Jorge Toledo El Colegio de la Frontera Sur (ECOSUR), Tapachula, Chiapas, México Pablo Montoya Programa Moscafrut SADER-SENASICA, Metapa, Chiapas, México Pablo Liedo El Colegio de la Frontera Sur (ECOSUR), Tapachula, Chiapas, México Walther Enkerlin Joint FAO/IAEA Programme of Nuclear Techniques in Food and Agriculture, Vienna, Austria Food and Agriculture Organization of the United Nations International Atomic Energy Agency Vienna, 2019 FOREWORD Effective fruit fly control requires an integrated pest management approach which may include the use of area-wide sterile insect technique (SIT). -

Integration of Entomopathogenic Fungi Into IPM Programs: Studies Involving Weevils (Coleoptera: Curculionoidea) Affecting Horticultural Crops
insects Review Integration of Entomopathogenic Fungi into IPM Programs: Studies Involving Weevils (Coleoptera: Curculionoidea) Affecting Horticultural Crops Kim Khuy Khun 1,2,* , Bree A. L. Wilson 2, Mark M. Stevens 3,4, Ruth K. Huwer 5 and Gavin J. Ash 2 1 Faculty of Agronomy, Royal University of Agriculture, P.O. Box 2696, Dangkor District, Phnom Penh, Cambodia 2 Centre for Crop Health, Institute for Life Sciences and the Environment, University of Southern Queensland, Toowoomba, Queensland 4350, Australia; [email protected] (B.A.L.W.); [email protected] (G.J.A.) 3 NSW Department of Primary Industries, Yanco Agricultural Institute, Yanco, New South Wales 2703, Australia; [email protected] 4 Graham Centre for Agricultural Innovation (NSW Department of Primary Industries and Charles Sturt University), Wagga Wagga, New South Wales 2650, Australia 5 NSW Department of Primary Industries, Wollongbar Primary Industries Institute, Wollongbar, New South Wales 2477, Australia; [email protected] * Correspondence: [email protected] or [email protected]; Tel.: +61-46-9731208 Received: 7 September 2020; Accepted: 21 September 2020; Published: 25 September 2020 Simple Summary: Horticultural crops are vulnerable to attack by many different weevil species. Fungal entomopathogens provide an attractive alternative to synthetic insecticides for weevil control because they pose a lesser risk to human health and the environment. This review summarises the available data on the performance of these entomopathogens when used against weevils in horticultural crops. We integrate these data with information on weevil biology, grouping species based on how their developmental stages utilise habitats in or on their hostplants, or in the soil. -

Entomopathogenic Fungi and Bacteria in a Veterinary Perspective
biology Review Entomopathogenic Fungi and Bacteria in a Veterinary Perspective Valentina Virginia Ebani 1,2,* and Francesca Mancianti 1,2 1 Department of Veterinary Sciences, University of Pisa, viale delle Piagge 2, 56124 Pisa, Italy; [email protected] 2 Interdepartmental Research Center “Nutraceuticals and Food for Health”, University of Pisa, via del Borghetto 80, 56124 Pisa, Italy * Correspondence: [email protected]; Tel.: +39-050-221-6968 Simple Summary: Several fungal species are well suited to control arthropods, being able to cause epizootic infection among them and most of them infect their host by direct penetration through the arthropod’s tegument. Most of organisms are related to the biological control of crop pests, but, more recently, have been applied to combat some livestock ectoparasites. Among the entomopathogenic bacteria, Bacillus thuringiensis, innocuous for humans, animals, and plants and isolated from different environments, showed the most relevant activity against arthropods. Its entomopathogenic property is related to the production of highly biodegradable proteins. Entomopathogenic fungi and bacteria are usually employed against agricultural pests, and some studies have focused on their use to control animal arthropods. However, risks of infections in animals and humans are possible; thus, further studies about their activity are necessary. Abstract: The present study aimed to review the papers dealing with the biological activity of fungi and bacteria against some mites and ticks of veterinary interest. In particular, the attention was turned to the research regarding acarid species, Dermanyssus gallinae and Psoroptes sp., which are the cause of severe threat in farm animals and, regarding ticks, also pets. -

Pathogenic and Enzyme Activities of the Entomopathogenic Fungus Tolypocladium Cylindrosporum (Ascomycota: Hypocreales) from Tierra Del Fuego, Argentina
Pathogenic and enzyme activities of the entomopathogenic fungus Tolypocladium cylindrosporum (Ascomycota: Hypocreales) from Tierra del Fuego, Argentina Ana C. Scorsetti1*, Lorena A. Elíades1, Sebastián A. Stenglein2, Marta N. Cabello1,3, Sebastián A. Pelizza1,4 & Mario C.N. Saparrat1,5,6 1. Instituto de Botánica Carlos Spegazzini (FCNyM-UNLP) 53 # 477, (1900), La Plata, Argentina; [email protected], [email protected] 2. Laboratorio de Biología Funcional y Biotecnología (BIOLAB)-CEBB-CONICET, Cátedra de Microbiología, Facultad de Agronomía de Azul, UNCPBA, República de Italia # 780, Azul (7300), Argentina; [email protected] 3. Comisión de Investigaciones Científicas de la provincia de Buenos Aires; [email protected] 4. Centro de Estudios Parasitológicos y de Vectores (CEPAVE), CCT-La Plata-CONICET-UNLP, Calle 2 # 584, La Plata (1900), Argentina; [email protected] 5. Instituto de Fisiología Vegetal (INFIVE), Universidad Nacional de La Plata (UNLP)- CCT-La Plata- Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Diag. 113 y 61, CC 327, 1900-La Plata, Argentina; [email protected] 6. Cátedra de Microbiología Agrícola, Facultad de Ciencias Agrarias y Forestales, UNLP, 60 y 119, 1900-La Plata, Argentina. * Corresponding author Received 27-IV-2011. Corrected 20-VIII-2011. Accepted 14-IX-2011. Abstract: Tolypocladium cylindrosporum is an entomopathogenic fungi that has been studied as a biological control agent against insects of several orders. The fungus has been isolated from the soil as well as from insects of the orders Coleoptera, Lepidoptera, Diptera and Hymenoptera. In this study, we analyzed the ability of a strain of T. cylindrosporum, isolated from soil samples taken in Tierra del Fuego, Argentina, to produce hydro- lytic enzymes, and to study the relationship of those activities to the fungus pathogenicity against pest aphids. -

Evaluation of Entomopathogenic Fungi (Ascomycota) for the Control of Cydia Pomonella (Lepidoptera: Tortricidae)
EVALUATION OF ENTOMOPATHOGENIC FUNGI (ASCOMYCOTA) FOR THE CONTROL OF CYDIA POMONELLA (LEPIDOPTERA: TORTRICIDAE) BY ASOMIBA RITA ABAAJEH A THESIS PRESENTED TO CAPE PENINSULA UNIVERSITY OF TECHNOLOGY IN FULFILMENT OF THE REQUIREMENTS FOR THE MASTERS OF TECHNOLOGY DEGREE IN HORTICULTURE. IN THE FACULTY OF APPLIED SCIENCES SUPERVISOR: DR NCHU, F. CO- SUPERVISOR: PROF LAUBSCHER, C. CAPE TOWN CAMPUS JANUARY 2014 i Title page EVALUATION OF ENTOMOPATHOGENIC FUNGI (ASCOMYCOTA) FOR THE CONTROL OF CYDIA POMONELLA (LEPIDOPTERA: TORTRICIDAE) ii DECLARATION I, Asomiba Rita Abaajeh, declare that the contents of this thesis represent my own unaided work, and that the thesis has not previously been submitted for academic examination towards any qualification. Furthermore, it represents my own opinions and not necessarily those of the Cape Peninsula University of Technology. Signed Date iii Dedication To God almighty for keeping me healthy and guiding me throughout this study To Dr. P. Igue To my parents; Mr & Mrs Abaajeh. iv _____________________________________________________________________________ ACKNOWLEDGEMENTS _____________________________________________________________________________ I want to thank: . My supervisor, Dr. Nchu, F. for the guidance and for being so passionate about entomopathogenic fungi. His knowledge of the subject helped me unlock the potentials that this subject holds. Dr. Karabu, S. for providing laboratory space for fungal isolation. Mr. Wohlfarter, M. for providing codling moth eggs and larvae used for this work and -

Effect of Beauveria Bassiana Fungal Infection on Survival and Feeding
Article Effect of Beauveria bassiana Fungal Infection on Survival and Feeding Behavior of Pine-Tree Lappet Moth (Dendrolimus pini L.) Marta Kovaˇc 1 , Nikola Lackovi´c 2 and Milan Pernek 1,* 1 Croatian Forest Research Institute, Cvjetno naselje 41, HR-10450 Jastrebarsko, Croatia; [email protected] 2 Arbofield Ltd., Mihanovi´ceva3, HR-10450 Jastrebarsko, Croatia; [email protected] * Correspondence: [email protected]; Tel.: +385-98-324-512 Received: 30 July 2020; Accepted: 4 September 2020; Published: 9 September 2020 Abstract: Research highlights: The pine-tree lappet moth, Dendrolimus pini, can cause serious needle defoliation on pines with outbreaks occurring over large geographical areas. Under laboratory conditions, the promising potential of the naturally occurring entomopathogenic fungus Beauveria bassiana was tested against D. pini larvae as a biological control method. Background and objectives: The aim of this study was to investigate the most effective concentration and treatment dose of B. bassiana conidial suspension and how it affected the survival and feeding behavior of the pest. Materials and methods: The first experiment applied the fungal suspension directly on the back of selected larvae, and in the second experiment, sporulating cadavers obtained in the first experiment were placed into Petri dishes with healthy individuals. Different doses per larvae [µL] and spore suspension concentration [spores/µL]) were used. The second experiment was designed to investigate the horizontal transmission of fungi by exposing individual caterpillars to a cadaver covered in B. bassiana mycelia. Mortality rates were analyzed by Chi-squared tests using absolute values for total mortality and B. bassiana- attributed mortality. The lethal time and feeding-disruption speed were analyzed with parametric and non-parametric tests with the aim to determine whether statistically significant differences were observed between treatments. -

Both LCCL-Domains of Human CRISPLD2 Have High Affinity for Lipid A
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimie 97 (2014) 66e71 Contents lists available at ScienceDirect Biochimie journal homepage: www.elsevier.com/locate/biochi Research paper Both LCCL-domains of human CRISPLD2 have high affinity for lipid A Viktor Vásárhelyi, Mária Trexler, László Patthy* Institute of Enzymology, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Budapest H-1518, PO Box 7, Hungary article info abstract Article history: The LCCL-domain is a recently defined protein module present in diverse extracellular multidomain Received 15 May 2013 proteins. Practically nothing is known about the molecular function of these domains; based on func- Accepted 20 September 2013 tional features of proteins harboring LCCL-domains it has been suggested that these domains might Available online 1 October 2013 function as lipopolysaccharide-binding domains. Here we show that the two LCCL-domains of human CRISPLD2 protein, a lipopolysaccharide-binding serum protein involved in defense against endotoxin Keywords: shock, have higher affinity for the lipid A, the toxic moiety of lipopolysaccharides than for ipopoly- CRISPLD2 protein saccharide. Our observation that the LCCL-domains of CRISPLD2 are specific for the toxic lipid A moiety of Endotoxin LCCL-domain the endotoxin suggests that it may block the interaction between endotoxins and the host endotoxin Lipid A receptors without interfering with the development of antibacterial immunity against the polysaccharide Lipopolysaccharide moiety of LPS. We suggest that the anti-inflammatory function of CRISPLD2 protein may account for its role in various pathological and developmental processes. Ó 2013 The Authors. -

MOLECULAR METHODS and ISOLATES of the ENTOMOPATHOGENIC FUNGUS Metarhizium Anisopliae for ENVIRONMENTALLY SUSTAINABLE CONTROL of GRASSHOPPERS in CANADA
MOLECULAR METHODS AND ISOLATES OF THE ENTOMOPATHOGENIC FUNGUS Metarhizium anisopliae FOR ENVIRONMENTALLY SUSTAINABLE CONTROL OF GRASSHOPPERS IN CANADA Susan Carol Entz B.Sc, University of Lethbridge, 1985 A Thesis Submitted to the School of Graduate Studies of the University of Lethbridge in Partial Fulfilment of the Requirements for the Degree MASTER OF SCIENCE Department of Geography (Environmental Science) University of Lethbridge LETHBRIDGE, ALBERTA, CANADA © Susan C. Entz, 2005 Abstract Metarhizium anisopliae var. acridum, a hyphomycetous fungus registered worldwide for grasshopper and locust control, is currently under consideration as a potential alternative to chemical insecticides for grasshopper control in Canada. Research in this thesis has contributed data required for the registration of biological control agents in Canada. A diagnostic PCR assay was developed for the specific detection of M. anisopliae var. acridum DNA. The assay was highly sensitive and effective for the detection of fungal DNA in infected grasshoppers. A survey of southern Alberta soils conducted in the spring of 2004 revealed the presence of Metarhizium spp. at low natural incidence. Two indigenous isolates demonstrated pathogenicity when bioassayed against laboratory-reared and field- collected grasshoppers. One of the isolates demonstrated virulence comparable to a commercial isolate. An analysis of historical weather data revealed that summer weather in the Prairie provinces should not preclude the efficacy of M. anisopliae var. acridum under local conditions. iii Preface The following thesis is presented partially in manuscript format. The introduction and literature review are combined in a single chapter (Chapter 1) in traditional format. Chapters 2, 3, and 4 are presented as manuscripts. References for Chapters 2 to 4 are combined in a general references section as outlined in the table of contents. -
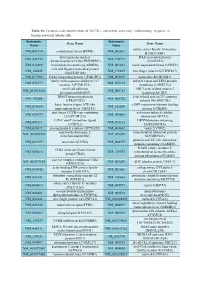
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

The Mechanisms of Social Immunity Against Fungal Infections in Eusocial Insects
toxins Review The Mechanisms of Social Immunity Against Fungal Infections in Eusocial Insects Long Liu 1,2, Xing-Ying Zhao 1, Qing-Bo Tang 2, Chao-Liang Lei 1 and Qiu-Ying Huang 1,* 1 Hubei Insect Resources Utilization and Sustainable Pest Management Key Laboratory, Huazhong Agricultural University, Wuhan 430070, China; [email protected] (L.L.); [email protected] (X.-Y.Z.); [email protected] (C.-L.L.) 2 Plant Protection College, Henan Agricultural University, Zhengzhou 450002, China; [email protected] * Correspondence: [email protected] Received: 29 March 2019; Accepted: 27 April 2019; Published: 29 April 2019 Abstract: Entomopathogenic fungus as well as their toxins is a natural threat surrounding social insect colonies. To defend against them, social insects have evolved a series of unique disease defenses at the colony level, which consists of behavioral and physiological adaptations. These colony-level defenses can reduce the infection and poisoning risk and improve the survival of societal members, and is known as social immunity. In this review, we discuss how social immunity enables the insect colony to avoid, resist and tolerate fungal pathogens. To understand the molecular basis of social immunity, we highlight several genetic elements and biochemical factors that drive the colony-level defense, which needs further verification. We discuss the chemosensory genes in regulating social behaviors, the antifungal secretions such as some insect venoms in external defense and the immune priming in internal defense. To conclude, we show the possible driving force of the fungal toxins for the evolution of social immunity. -

Analysis of the Plasmodium Falciparum Proteome by High-Accuracy Mass Organism
CORE Metadata, citation and similar papers at core.ac.uk Provided by University of Southern Denmark Research Output letters to nature 12. Lasonder, E. et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass organism. Here we describe a large-scale, high-accuracy (average spectrometry. Nature 419, 537–542 (2002). 13. Ewing, B. & Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. deviation less than 0.02 Da at 1,000 Da) mass spectrometric 1–3 Genome Res. 8, 186–194 (1998). proteome analysis of selected stages of the human malaria 14. Oefner, P. J. et al. Efficient random subcloning of DNA sheared in a recirculating point-sink flow parasite Plasmodium falciparum. The analysis revealed 1,289 system. Nucleic Acids Res. 24, 3879–3886 (1996). proteins of which 714 proteins were identified in asexual blood 15. Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998). stages, 931 in gametocytes and 645 in gametes. The last two 16. Gordon, D., Abajian, C. & Green, P. Consed: a graphical tool for sequence finishing. Genome Res. 8, groups provide insights into the biology of the sexual stages of 195–202 (1998). the parasite, and include conserved, stage-specific, secreted and 17. Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. membrane-associated proteins. A subset of these proteins con- J. Mol. Biol. 215, 403–410 (1990). 18. Apweiler, R. et al. The InterPro database, an integrated documentation resource for protein families, tain domains that indicate a role in cell–cell interactions, and domains and functional sites. -

The Development of Immunity in a Social Insect: Evidence for the Group Facilitation of Disease Resistance
The development of immunity in a social insect: Evidence for the group facilitation of disease resistance James F. A. Traniello*, Rebeca B. Rosengaus, and Keely Savoie Department of Biology, Boston University, 5 Cummington Street, Boston, MA 02215 Communicated by Bert Ho¨lldobler, University of Wurzburg, Wurzburg, Germany, March 25, 2002 (received for review October 31, 2001) The extraordinary diversity and ecological success of the social absent in naı¨ve termites. The dynamics of the immune response insects has been attributed to their ability to cope with the rich and of Z. angusticollis and its associated hemolymph protein profile often infectious microbial community inhabiting their nests and resemble immunization-related protective changes in the protein feeding sites. Mechanisms of disease control used by eusocial constituents of the phylogenetically related roaches (17). species include antibiotic glandular secretions, mutual grooming, Here we show that the level of immunocompetence attained removal of diseased individuals from the nest, and the innate and by termites depends on association with colony members and adaptive immune responses of colony members. Here we demon- that the disease resistance of individuals that have not experi- strate that after a challenge exposure to the entomopathogenic enced direct contact with a pathogen can be significantly en- fungus Metarhizium anisopliae, dampwood termites Zootermopsis hanced through interactions with immunized nestmates. Our angusticollis have higher survivorship when individuals develop studies of the development of immunity in Z. angustocollis reveal immunity as group members. Furthermore, termites significantly novel social mechanisms of infection control. improve their ability to resist infection when they are placed in contact with previously immunized nestmates.