Parophrys Vetulus) Across an Estuarine Gradient of Contaminants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Winter Flounder Pseudopleuronectes Americanus Stock Enhancement in New Hampshire: Developing Optimal Release Strategies
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Spring 2002 Winter flounder Pseudopleuronectes americanus stock enhancement in New Hampshire: Developing optimal release strategies Elizabeth Alden Fairchild University of New Hampshire, Durham Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation Fairchild, Elizabeth Alden, "Winter flounder Pseudopleuronectes americanus stock enhancement in New Hampshire: Developing optimal release strategies" (2002). Doctoral Dissertations. 62. https://scholars.unh.edu/dissertation/62 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Disease List for Aquaculture Health Certificate
Quarantine Standard for Designated Species of Imported/Exported Aquatic Animals [Attached Table] 4. Listed Diseases & Quarantine Standard for Designated Species Listed disease designated species standard Common name Disease Pathogen 1. Epizootic haematopoietic Epizootic Perca fluviatilis Redfin perch necrosis(EHN) haematopoietic Oncorhynchus mykiss Rainbow trout necrosis virus(EHNV) Macquaria australasica Macquarie perch Bidyanus bidyanus Silver perch Gambusia affinis Mosquito fish Galaxias olidus Mountain galaxias Negative Maccullochella peelii Murray cod Salmo salar Atlantic salmon Ameirus melas Black bullhead Esox lucius Pike 2. Spring viraemia of Spring viraemia of Cyprinus carpio Common carp carp, (SVC) carp virus(SVCV) Grass carp, Ctenopharyngodon idella white amur Hypophthalmichthys molitrix Silver carp Hypophthalmichthys nobilis Bighead carp Carassius carassius Crucian carp Carassius auratus Goldfish Tinca tinca Tench Sheatfish, Silurus glanis European catfish, wels Negative Leuciscus idus Orfe Rutilus rutilus Roach Danio rerio Zebrafish Esox lucius Northern pike Poecilia reticulata Guppy Lepomis gibbosus Pumpkinseed Oncorhynchus mykiss Rainbow trout Abramis brama Freshwater bream Notemigonus cysoleucas Golden shiner 3.Viral haemorrhagic Viral haemorrhagic Oncorhynchus spp. Pacific salmon septicaemia(VHS) septicaemia Oncorhynchus mykiss Rainbow trout virus(VHSV) Gadus macrocephalus Pacific cod Aulorhynchus flavidus Tubesnout Cymatogaster aggregata Shiner perch Ammodytes hexapterus Pacific sandlance Merluccius productus Pacific -
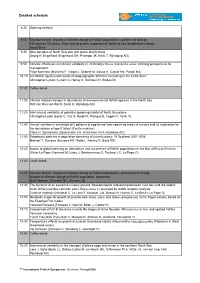
Schedule Onlinepdf
Detailed schedule 8:30 Opening session 9:00 Keynote lecture: Impacts of climate change on flatfish populations - patterns of change 100 days to 100 years: Short and long-term responses of flatfish to sea temperature change David Sims 9:30 Nine decades of North Sea sole and plaice distributions Georg H. Engelhard (Engelhard GH, Pinnegar JK, Kell LT, Rijnsdorp AD) 9:50 Climatic effects on recruitment variability in Platichthys flesus and Solea solea: defining perspectives for management. Filipe Martinho (Martinho F, Viegas I, Dolbeth M, Sousa H, Cabral HN, Pardal MA) 10:10 Are flatfish species with southern biogeographic affinities increasing in the Celtic Sea? Christopher Lynam (Lynam C, Harlay X, Gerritsen H, Stokes D) 10:30 Coffee break 11:00 Climate related changes in abundance of non-commercial flatfish species in the North Sea Ralf van Hal (van Hal R, Smits K, Rijnsdorp AD) 11:20 Inter-annual variability of potential spawning habitat of North Sea plaice Christophe Loots (Loots C, Vaz S, Koubii P, Planque B, Coppin F, Verin Y) 11:40 Annual variation in simulated drift patterns of egg/larvae from spawning areas to nursery and its implication for the abundance of age-0 turbot (Psetta maxima) Claus R. Sparrevohn (Sparrevohn CR, Hinrichsen H-H, Rijnsdorp AD) 12:00 Broadscale patterns in population dynamics of juvenile plaice: W Scotland 2001-2008 Michael T. Burrows (Burrows MT, Robb L, Harvey R, Batty RS) 12:20 Impact of global warming on abundance and occurrence of flatfish populations in the Bay of Biscay (France) Olivier Le Pape (Hermant -

NHBSS 034 1G Wongratana R
NAT. NAT. HIST. BULL. SIAM So c. 34 (1) :65 ・70 ,1986 RECORD OF AMBICOLORATION IN CYNOGLOSSUS (PISCES : CYNOGLOSSIDAE) FROM THAILAND Thosaporn Thosaporn Wongratana * ABSTRACT An almost ambico10rate “Four- Ii ned tongu e- sole" ,Cynoglossus bilinealus (La cepede) , is is reported from Thailand. It is presumab1y the first record for the genus. Except for most of of the head on the blind side and its.corresponding finrays ,which are pale as in normal specimens ,the body and fins are pigmented. The norma Jl y cycloid scales on the blind side in in the pigmented area are who Jl y replaced by ctenoid scales ,but those on the unpigmented part part on the head are cyc10id. The latera1line sca1es of the pigmented area on the blind side are are cycloid. The pelvic fins are entirely separated from the anal fin by the absence of membrane. membrane. No other major externa1 anomaly is found. PREVIOUS ACCOUNTS Abnormalities Abnormalities in coloration 釘 e more common among members of the order Pleuronectiformes Pleuronectiformes than in any other group of fishes. Other anomalies occasionally in those those fishes are a hooked dorsal fin , incomplete eye migration and side reversa l. Abnormal pigmentation in flatfish is divided into three main types: ambicoloration , albinism ,and xanthochromism (DEVEEN , 1969; COLMAN ,1972 勾). P 町 “tia 剖10 町Ir incomplete ambic ∞010 町r羽 ion is mor 問ec ∞ommon than trunk pigmentation , nearly complete amb 凶ic ∞0- lor 問at “ion and complete ambic ∞010 町ra 創ti 拘on (υJONES & MENON , 1950). NORMAN'S (1 934) previous previous explanation of this phenomenon ,later accepted by many authors ,was that “..訓 nbicoloration merely represents variation in the direction of the original bilateral symmetrical symmetrical condition of the ancester of flatfishes." It is also regularly observed that that wholly ambicolorate fishes normally display a higher degree of symmetry than normal normal specimens in pigmentation ,scales , paired fins and final position of the eyes (NORMAN , 1934; COLMAN , 1972). -

Pleuronectidae
FAMILY Pleuronectidae Rafinesque, 1815 - righteye flounders [=Heterosomes, Pleronetti, Pleuronectia, Diplochiria, Poissons plats, Leptosomata, Diprosopa, Asymmetrici, Platessoideae, Hippoglossoidinae, Psettichthyini, Isopsettini] Notes: Hétérosomes Duméril, 1805:132 [ref. 1151] (family) ? Pleuronectes [latinized to Heterosomi by Jarocki 1822:133, 284 [ref. 4984]; no stem of the type genus, not available, Article 11.7.1.1] Pleronetti Rafinesque, 1810b:14 [ref. 3595] (ordine) ? Pleuronectes [published not in latinized form before 1900; not available, Article 11.7.2] Pleuronectia Rafinesque, 1815:83 [ref. 3584] (family) Pleuronectes [senior objective synonym of Platessoideae Richardson, 1836; family name sometimes seen as Pleuronectiidae] Diplochiria Rafinesque, 1815:83 [ref. 3584] (subfamily) ? Pleuronectes [no stem of the type genus, not available, Article 11.7.1.1] Poissons plats Cuvier, 1816:218 [ref. 993] (family) Pleuronectes [no stem of the type genus, not available, Article 11.7.1.1] Leptosomata Goldfuss, 1820:VIII, 72 [ref. 1829] (family) ? Pleuronectes [no stem of the type genus, not available, Article 11.7.1.1] Diprosopa Latreille, 1825:126 [ref. 31889] (family) Platessa [no stem of the type genus, not available, Article 11.7.1.1] Asymmetrici Minding, 1832:VI, 89 [ref. 3022] (family) ? Pleuronectes [no stem of the type genus, not available, Article 11.7.1.1] Platessoideae Richardson, 1836:255 [ref. 3731] (family) Platessa [junior objective synonym of Pleuronectia Rafinesque, 1815, invalid, Article 61.3.2 Hippoglossoidinae Cooper & Chapleau, 1998:696, 706 [ref. 26711] (subfamily) Hippoglossoides Psettichthyini Cooper & Chapleau, 1998:708 [ref. 26711] (tribe) Psettichthys Isopsettini Cooper & Chapleau, 1998:709 [ref. 26711] (tribe) Isopsetta SUBFAMILY Atheresthinae Vinnikov et al., 2018 - righteye flounders GENUS Atheresthes Jordan & Gilbert, 1880 - righteye flounders [=Atheresthes Jordan [D. -

(Paralichthys Lethostigma) in the Galveston Bay Estuary, TX
DISTRIBUTION, CONDITION, AND GROWTH OF NEWLY SETTLED SOUTHERN FLOUNDER (Paralichthys lethostigma) IN THE GALVESTON BAY ESTUARY, TX A Thesis by LINDSAY ANN GLASS Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE May 2006 Major Subject: Wildlife and Fisheries Sciences DISTRIBUTION, CONDITION, AND GROWTH OF NEWLY SETTLED SOUTHERN FLOUNDER (Paralichthys lethostigma) IN THE GALVESTON BAY ESTUARY, TX A Thesis by LINDSAY ANN GLASS Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Approved by: Chair of Committee, Jay R. Rooker Committee Members, William H. Neill Antonietta Quigg Head of Department, Delbert M.Gatlin III May 2006 Major Subject: Wildlife and Fisheries Sciences iii ABSTRACT Distribution, Condition, and Growth of Newly Settled Southern Flounder (Paralichthys lethostigma) in the Galveston Bay Estuary, TX. (May 2006) Lindsay Ann Glass, B.S., Texas A&M University-Galveston Chair of Advisory Committee: Dr. Jay R. Rooker Several flatfish species including southern flounder (Paralichthys lethostigma) recruit to estuaries during early life. Therefore, the evaluation of estuarine sites and habitats that serve as nurseries is critical to conservation and management efforts. I used biochemical condition and growth measurements in conjunction with catch-density data to evaluate settlement sites used by southern flounder in the Galveston Bay Estuary (GBE). In 2005, beam-trawl collections were made in three major sections of the GBE (East Bay, West Bay, Galveston Bay), and three sites were sampled in each bay. -

Yellowfin Trawling Fish Images 2013 09 16
Fishes captured aboard the RV Yellowfin in otter trawls: September 2013 Order: Aulopiformes Family: Synodontidae Species: Synodus lucioceps common name: California lizardfish Order: Gadiformes Family: Merlucciidae Species: Merluccius productus common name: Pacific hake Order: Ophidiiformes Family: Ophidiidae Species: Chilara taylori common name: spotted cusk-eel plainfin specklefin Order: Batrachoidiformes Family: Batrachoididae Species: Porichthys notatus & P. myriaster common name: plainfin & specklefin midshipman plainfin specklefin Order: Batrachoidiformes Family: Batrachoididae Species: Porichthys notatus & P. myriaster common name: plainfin & specklefin midshipman plainfin specklefin Order: Batrachoidiformes Family: Batrachoididae Species: Porichthys notatus & P. myriaster common name: plainfin & specklefin midshipman Order: Gasterosteiformes Family: Syngnathidae Species: Syngnathus leptorynchus common name: bay pipefish Order: Scorpaeniformes Family: Scorpaenidae Species: Sebastes semicinctus common name: halfbanded rockfish Order: Scorpaeniformes Family: Scorpaenidae Species: Sebastes dalli common name: calico rockfish Order: Scorpaeniformes Family: Scorpaenidae Species: Sebastes saxicola common name: stripetail rockfish Order: Scorpaeniformes Family: Scorpaenidae Species: Sebastes diploproa common name: splitnose rockfish Order: Scorpaeniformes Family: Scorpaenidae Species: Sebastes rosenblatti common name: greenblotched rockfish juvenile Order: Scorpaeniformes Family: Scorpaenidae Species: Sebastes levis common name: cowcod Order: -

Distribution and Abundance of Pleuronectiformes Larvae Off Southeastern Brazil
BRAZILIAN JOURNAL OF OCEANOGRAPHY, 62(1):23-34, 2014 DISTRIBUTION AND ABUNDANCE OF PLEURONECTIFORMES LARVAE OFF SOUTHEASTERN BRAZIL Camilla Nunes Garbini*, Maria de Lourdes Zani-Teixeira , Márcio Hidekazu Ohkawara and Mario Katsuragawa Instituto Oceanográfico da Universidade de São Paulo (Praça do Oceanográfico, 191, 05508-120 São Paulo, SP, Brasil) *Corresponding author: [email protected] http://dx.doi.org/10.1590/S1679-87592014051706201 ABSTRACT The objective of this study was the description of the composition, abundance and density in horizontal and vertical distribution of Pleuronectiformes larvae on the southeastern Brazilian continental shelf. The samples were collected with bongo nets and a Multi Plankton Sampler (MPS), both in summer and winter 2002. A total of 352 flatfishes larvae were collected in summer and 343 in winter, representing three families and a total of 13 taxa: Paralichthyidae ( Citharichthys cornutus, C. spilopterus, Citharichthys sp ., Cyclopsetta chittendeni, Syacium spp ., Etropus spp . and Paralichthys spp .), Bothidae ( Bothus ocellatus and Monolene antillarum ) and Cynoglossidae ( Symphurus trewavasae, S. jenynsi, S. plagusia and S. ginsburgi ). The most abundant taxa were Etropus spp ., Syacium spp . and Bothus ocellatus . Etropus spp . occurred mainly as far out as the 200 m isobath and Syacium spp . from 100 m. B. ocellatus was present mainly in the oceanic zone between Ubatuba and Rio de Janeiro as from the 200 m isobath. The greatest average densities of these species occurred in the strata from 0 to 20 m depth in summer and between 20 and 40 m in winter. RESUMO O objetivo deste estudo foi descrever a composição, abundância, densidade, distribuição horizontal e vertical das larvas de Pleuronectiformes ao longo da plataforma continental Sudeste brasileira. -

Using Stable Isotopes to Discern Mechanisms of Connectivity in Estuarine Detritus-Based Food Webs
Vol. 518: 13–29, 2015 MARINE ECOLOGY PROGRESS SERIES Published January 7 doi: 10.3354/meps11066 Mar Ecol Prog Ser Using stable isotopes to discern mechanisms of connectivity in estuarine detritus-based food webs Emily R. Howe*, Charles A. Simenstad University of Washington, School of Aquatic and Fishery Sciences, Seattle, WA 98195-5020, USA ABSTRACT: In this paper, we focus on 2 mechanisms of cross-boundary food web connectivity in Puget Sound estuaries: passive transport of water-advected organic matter (OM) and active move- ment of organisms. Both mechanisms serve as potential vectors of food web connectivity, but little research has investigated whether landscape setting changes the dominance of one mechanism over another, or whether the influence of organism movement on food web connectivity can be detected in estuarine systems. We use fish diets, stable isotopes and Bayesian mixing models to identify differences in OM sources assimilated by estuarine fishes, testing whether increased organism mobility or increased fluvial influence results in greater food web connectivity. We com- pare food web connectivity in 2 different estuaries, one displaying limited freshwater inputs, and the other the terminus of a major river system. Within each estuary, we investigate whether differ- ences in behavioral life history traits correspond to differences in the diets, isotopic signatures and OM assimilation of 2 fish species: bay pipefish Syngnathus leptorhynchus, which displays site fidelity to eelgrass beds, and the more transitory juvenile English sole Parophrys vetulus, which moves throughout estuarine deltas during the early demersal growth stage. Our results show water advection plays a dominant role in large-scale OM transport and delivery to adjoining eco- systems in the fluvial estuary, while organism movement provides the more important mechanism of food web connectivity in the estuary exhibiting minor fluvial discharge. -

Locus Number Estimation of MHC Class II B in Stone Flounder and Japanese Flounder
Int. J. Mol. Sci. 2015, 16, 6000-6017; doi:10.3390/ijms16036000 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Locus Number Estimation of MHC Class II B in Stone Flounder and Japanese Flounder Jiajun Jiang †, Chunmei Li †, Quanqi Zhang and Xubo Wang * Key Laboratory of Marine Genetics and Breeding (Ocean University of China), Ministry of Education, Qingdao 266003, China; E-Mails: [email protected] (J.J.); [email protected] (C.L.); [email protected] (Q.Z.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel./Fax: +86-532-8203-1986. Academic Editor: Ritva Tikkanen Received: 2 December 2014 / Accepted: 25 December 2014 / Published: 13 March 2015 Abstract: Members of major histocompatibility complex (MHC) family are important in immune systems. Great efforts have been made to reveal their complicated gene structures. But many existing studies focus on partial sequences of MHC genes. In this study, by gene cloning and sequencing, we identified cDNA sequences and DNA sequences of the MHC class II B in two flatfishes, stone flounder (Kareius bicoloratus) and homozygous diploid Japanese flounder (Paralichthys olivaceus). Eleven cDNA sequences were acquired from eight stone flounder individuals, and most of the polymorphic sites distributed in exons 2 and 3. Twenty-eight alleles were identified from the DNA fragments in these eight individuals. It could be deduced from their Bayesian inference phylogenetic tree that at least four loci of MHC class II B exist in stone flounder. The detailed whole-length DNA sequences in one individual were analyzed, revealing that the intron length varied among different loci. -
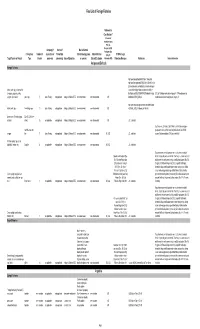
2018 Final LOFF W/ Ref and Detailed Info
Final List of Foreign Fisheries Rationale for Classification ** (Presence of mortality or injury (P/A), Co- Occurrence (C/O), Company (if Source of Marine Mammal Analogous Gear Fishery/Gear Number of aquaculture or Product (for Interactions (by group Marine Mammal (A/G), No RFMO or Legal Target Species or Product Type Vessels processor) processing) Area of Operation or species) Bycatch Estimates Information (N/I)) Protection Measures References Detailed Information Antigua and Barbuda Exempt Fisheries http://www.fao.org/fi/oldsite/FCP/en/ATG/body.htm http://www.fao.org/docrep/006/y5402e/y5402e06.htm,ht tp://www.tradeboss.com/default.cgi/action/viewcompan lobster, rock, spiny, demersal fish ies/searchterm/spiny+lobster/searchtermcondition/1/ , (snappers, groupers, grunts, ftp://ftp.fao.org/fi/DOCUMENT/IPOAS/national/Antigua U.S. LoF Caribbean spiny lobster trap/ pot >197 None documented, surgeonfish), flounder pots, traps 74 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented A/G AndBarbuda/NPOA_IUU.pdf Caribbean mixed species trap/pot are category III http://www.nmfs.noaa.gov/pr/interactions/fisheries/tabl lobster, rock, spiny free diving, loops 19 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented A/G e2/Atlantic_GOM_Caribbean_shellfish.html Queen conch (Strombus gigas), Dive (SCUBA & free molluscs diving) 25 not applicable not applicable Antigua & Barbuda EEZ none documented none documented A/G U.S. trade data Southeastern U.S. Atlantic, Gulf of Mexico, and Caribbean snapper- handline, hook and grouper and other reef fish bottom longline/hook-and-line/ >5,000 snapper line 71 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented N/I, A/G U.S. -

Bilateral Asymmetry and Bilateral Variation in Fishes *
BILATERAL ASYMMETRY AND BILATERAL VARIATION IN FISHES * bARL L. HUBBS AND LAURA C. HUBBS CONTENTS PAGE Introduction ................................................................................................................... 230 Statistical methods ....................................................................................................... 231 Dextrality and sinistrality in flatfishes .................................................................. 234 Reversal of sides in flounders .............................................................................. 236 Decreased viability of reversed flounders ......................................................... 240 Incomplete mirror imaging in reversed flounders .......................................... 243 A completely reversed flatfish .............................................................................. 245 Interpretation of reversal in flatfishes ............................................................... 246 Teratological return toward symmetry ............................................................. 249 Secondary asymmetries in flatfishes .................................................................... 250 Bilateral variation in number of rays in paired fins on the two sides of flatfishes ................................................................................................................. 254 Asymmetries and bilateral variations in essentially symmetrical fishes ....... 263 Bilateral variation in number of rays in the left