Function-Informed Transcriptome Analysis of Drosophila Renal Tubule
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Classification of Esterases: an Important Gene Family Involved in Insecticide Resistance - a Review
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol. 107(4): 437-449, June 2012 437 The classification of esterases: an important gene family involved in insecticide resistance - A Review Isabela Reis Montella1,2, Renata Schama1,2,3/+, Denise Valle1,2,3 1Laboratório de Fisiologia e Controle de Artrópodes Vetores, Instituto Oswaldo Cruz-Fiocruz, Av. Brasil 4365, 21040-900 Rio de Janeiro, RJ, Brasil 2Instituto de Biologia do Exército, Rio de Janeiro, RJ, Brasil 3Instituto Nacional de Ciência e Tecnologia em Entomologia Molecular, Rio de Janeiro, RJ, Brasil The use of chemical insecticides continues to play a major role in the control of disease vector populations, which is leading to the global dissemination of insecticide resistance. A greater capacity to detoxify insecticides, due to an increase in the expression or activity of three major enzyme families, also known as metabolic resistance, is one major resistance mechanisms. The esterase family of enzymes hydrolyse ester bonds, which are present in a wide range of insecticides; therefore, these enzymes may be involved in resistance to the main chemicals employed in control programs. Historically, insecticide resistance has driven research on insect esterases and schemes for their classification. Currently, several different nomenclatures are used to describe the esterases of distinct species and a universal standard classification does not exist. The esterase gene family appears to be rapidly evolving and each insect species has a unique complement of detoxification genes with only a few orthologues across species. The examples listed in this review cover different aspects of their biochemical nature. However, they do not appear to contribute to reliably distinguish among the different resistance mechanisms. -

Interactions of Zinc with the Intestinal Epithelium - Effects On
Aus dem Institut für Veterinär-Physiologie des Fachbereichs Veterinärmedizin der Freien Universität Berlin Interactions of zinc with the intestinal epithelium - effects on transport properties and zinc homeostasis Inaugural-Dissertation zur Erlangung des Grades eines Doktors der Veterinärmedizin an der Freien U niversität Berlin vorgelegt von Eva-Maria Näser, geb. Gefeller Tierärztin aus Kassel Berlin 2015 Journal-Nr.: 3813 Gefördert durch die Deutsche Forschungsgemeinschaft und die H.W. Schaumann Stiftung Gedruckt mit Genehmigung des Fachbereichs Veterinärmedizin der Freien Universität Berlin Dekan: Univ.-Prof. Dr. Jürgen Zentek Erster Gutachter: Univ.-Prof. Dr. Jörg Rudolf Aschenbach Zweiter Gutachter: Prof. Dr. Holger Martens Dritter Gutachter: Prof. Dr. Robert Klopfleisch Deskriptoren (nach CAB-Thesaurus): pigs, weaning, zinc, intestines, epithelium, jejunum, ion transport Tag der Promotion: 15.09.2015 Bibliografische Information der Deutschen Nationalbibliothek Die Deutsche Nationalbibliothek verzeichnet diese Publikation in der Deutschen Nationalbibliografie; detaillierte bibliografische Daten sind im Internet über <http://dnb.ddb.de> abrufbar. ISBN: 978-3-86387-656-2 Zugl.: Berlin, Freie Univ., Diss., 2015 Dissertation, Freie Universität Berlin D 188 Dieses Werk ist urheberrechtlich geschützt. Alle Rechte, auch die der Übersetzung, des Nachdruckes und der Vervielfältigung des Buches, oder Teilen daraus, vorbehalten. Kein Teil des Werkes darf ohne schriftliche Genehmigung des Verlages in irgendeiner Form reproduziert oder unter Verwendung elektronischer Systeme verarbeitet, vervielfältigt oder verbreitet werden. Die Wiedergabe von Gebrauchsnamen, Warenbezeichnungen, usw. in diesem Werk berechtigt auch ohne besondere Kennzeichnung nicht zu der Annahme, dass solche Namen im Sinne der Warenzeichen- und Markenschutz-Gesetzgebung als frei zu betrachten wären und daher von jedermann benutzt werden dürfen. This document is protected by copyright law. -

Supporting Information
Supporting Information Figure S1. The functionality of the tagged Arp6 and Swr1 was confirmed by monitoring cell growth and sensitivity to hydeoxyurea (HU). Five-fold serial dilutions of each strain were plated on YPD with or without 50 mM HU and incubated at 30°C or 37°C for 3 days. Figure S2. Localization of Arp6 and Swr1 on chromosome 3. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 3L, Tel 3R, CEN3, and the RP gene are shown under the panels. Figure S3. Localization of Arp6 and Swr1 on chromosome 4. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) in the whole chromosome region are compared. The position of Tel 4L, Tel 4R, CEN4, SWR1, and RP genes are shown under the panels. Figure S4. Localization of Arp6 and Swr1 on the region including the SWR1 gene of chromosome 4. The binding of Arp6- FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position and orientation of the SWR1 gene is shown. Figure S5. Localization of Arp6 and Swr1 on chromosome 5. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 5L, Tel 5R, CEN5, and the RP genes are shown under the panels. Figure S6. Preferential localization of Arp6 and Swr1 in the 5′ end of genes. Vertical bars represent the binding ratio of proteins in each locus. -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Enzymatic Degradation of Organophosphorus Pesticides and Nerve Agents by EC: 3.1.8.2
catalysts Review Enzymatic Degradation of Organophosphorus Pesticides and Nerve Agents by EC: 3.1.8.2 Marek Matula 1, Tomas Kucera 1 , Ondrej Soukup 1,2 and Jaroslav Pejchal 1,* 1 Department of Toxicology and Military Pharmacy, Faculty of Military Health Sciences, University of Defence, Trebesska 1575, 500 01 Hradec Kralove, Czech Republic; [email protected] (M.M.); [email protected] (T.K.); [email protected] (O.S.) 2 Biomedical Research Center, University Hospital Hradec Kralove, Sokolovska 581, 500 05 Hradec Kralove, Czech Republic * Correspondence: [email protected] Received: 26 October 2020; Accepted: 20 November 2020; Published: 24 November 2020 Abstract: The organophosphorus substances, including pesticides and nerve agents (NAs), represent highly toxic compounds. Standard decontamination procedures place a heavy burden on the environment. Given their continued utilization or existence, considerable efforts are being made to develop environmentally friendly methods of decontamination and medical countermeasures against their intoxication. Enzymes can offer both environmental and medical applications. One of the most promising enzymes cleaving organophosphorus compounds is the enzyme with enzyme commission number (EC): 3.1.8.2, called diisopropyl fluorophosphatase (DFPase) or organophosphorus acid anhydrolase from Loligo Vulgaris or Alteromonas sp. JD6.5, respectively. Structure, mechanisms of action and substrate profiles are described for both enzymes. Wild-type (WT) enzymes have a catalytic activity against organophosphorus compounds, including G-type nerve agents. Their stereochemical preference aims their activity towards less toxic enantiomers of the chiral phosphorus center found in most chemical warfare agents. Site-direct mutagenesis has systematically improved the active site of the enzyme. These efforts have resulted in the improvement of catalytic activity and have led to the identification of variants that are more effective at detoxifying both G-type and V-type nerve agents. -

( 12 ) United States Patent
US007459539B2 ( 12) United States Patent ( 10 ) Patent No. : US 7 , 459 ,539 B2 Challita - Eid et al. (45 ) Date of Patent: Dec. 2 , 2008 ( 54 ) ANTIBODY THAT BINDS ZINC WO WO 03050236 6 / 2003 TRANSPORTER PROTEIN 108P5H8 ( 75 ) Inventors : Pia M . Challita - Eid , Encino , CA (US ) ; OTHER PUBLICATIONS Mary Faris , Los Angeles , CA (US ) ; Murgia et al . Cloning, expression , and vesicular localization of zinc Daniel E . H . Afar , Brisbane , CA (US ) ; transporter Dri 27 /ZnT4 in intestinal tissue and cells . Am J Physiol Rene S . Hubert , Los Angeles , CA (US ) ; 277 (Gastrointest Liver Physiol 40 ) : G1231 -61239 , 1999 . * Steve Chappell Mitchell, Gurnee , IL Kaufman et al. Blood 9 : 3178 -3184 , 1999 . * (US ) ; Elana Levin , Los Angeles, CA Wang et al . Rapid analysis of gene expression (RAGE ) facilitates (US ) ; Karen Jane Meyrick Morrison , universal expression profiling . Nucleic Acids Res 27 (23 ) : 4609 4618 , 1999 . * Santa Monica , CA (US ) ; Arthur B . Campbell et al. Totipotency of multipotentiality of cultured cells : Raitano , Los Angeles , CA (US ) ; Aya applications and progress . Theriogenology 47 : 63 -72 , 1997 . * Jakobovits , Beverly Hills , CA (US ) Moore , G . Genetically engineered antibodies. Clin Chem 35 ( 9 ) : 1849 - 1853 , 1989 . * (73 ) Assignee : Agensys, Inc ., Santa Monica , CA (US ) Dillman et al .Monoclonal antibodies in the treatment ofmalignancy : basic concepts and recent developments . Cancer Invest 19 ( 8 ) : 833 ( * ) Notice: Subject to any disclaimer, the term of this 841, 2001. * patent is extended or adjusted under 35 Skolnick et al. From genes to protein structure and function : novel U . S . C . 154 ( b ) by 1273 days . applications of computational approaches in the genomic era . -
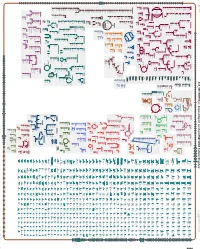
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

Reconsideration of the Catalytic Center and Mechanism of Mammalian
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 7187-7191, August 1995 Genetics Reconsideration of the catalytic center and mechanism of mammalian paraoxonase/arylesterase (organophosphatase/A-esterase/site-directed mutagenesis/high density lipoprotein/phosphotriesterase) ROBERT C. SORENSON*, SERGIO L. PRIMO-PARMOt, CHUNG-LIANG Kuot, STEVE ADKINS0§, OKSANA LOCKRIDGEI, AND BERT N. LA DU*tll *Department of Pharmacology, University of Michigan Medical School, Ann Arbor, MI 48109-0632; tDepartment of Anesthesiology, Research Division, 4038 Kresge Research II, University of Michigan Medical School, Ann Arbor, MI 48109-0572; tDepartment of Environmental and Industrial Health, School of Public Health, University of Michigan, Ann Arbor, MI 48109-2029; §Mental Health Research Institute and Department of Human Genetics, University of Michigan Medical School, Ann Arbor, MI 48109-0720; and lEppley Institute, University of Nebraska Medical Center, Omaha, NB 68198-6805 Communicated by James V Neel, University ofMichigan Medical School, Ann Arbor, MI, May 2, 1995 ABSTRACT For three decades, mammalian paraoxonase basis for the human polymorphism was the presence of argi- (A-esterase, aromatic esterase, arylesterase; PON, EC 3.1.8.1) nine (high PON activity, B-type allozyme) or glutamine (low has been thought to be a cysteine esterase demonstrating PON activity, A-type allozyme) at position 191 (11-13). structural and mechanistic homologies with the serine ester- Augustinsson (14, 15) suggested that PON, the carboxyes- ases (cholinesterases and carboxyesterases). Human, mouse, terases, and the cholinesterases were products of divergent and rabbit PONs each contain only three cysteine residues, evolution from an ancestral arylesterase but that PON utilized and their positions within PON have been conserved. -

Electronic Supplementary Material (ESI) for Analytical Methods. This Journal Is © the Royal Society of Chemistry 2014
Electronic Supplementary Material (ESI) for Analytical Methods. This journal is © The Royal Society of Chemistry 2014 Table S2. List of proteins detected in HCC serum. # # MW Accession Coverage Pepti calc. pI Score Description PSMs [kDa] des gi105990532 25.03 1012 66 515.2 7.05 1343.34 apolipoprotein B-100 precursor [Homo sapiens] gi225311 25.31 977 66 512.6 6.98 1324.72 lipoprotein B100 gi225310 24.57 972 65 515.2 7.06 1310.41 lipoprotein B100 gi119370332 52.25 1738 59 187.0 6.40 1995.72 Complement C3 gi122920512 76.07 4583 49 66.4 5.87 8935.61 Chain A, Human Serum Albumin Complexed With Myristate And Aspirin gi55669910 75.52 4500 46 65.2 5.80 8730.74 Chain A, Crystal Structure Of The Ga Module Complexed With Human Serum Albumin gi308153640 36.77 1109 35 163.2 6.46 1690.96 RecName: Full=Alpha-2-macroglobulin; Short=Alpha-2-M; AltName: Full=C3 and PZP- like alpha-2-macroglobulin domain-containing protein 5; Flags: Precursor gi66932947 36.77 1103 35 163.2 6.42 1690.30 alpha-2-macroglobulin precursor [Homo sapiens] gi118138017 49.95 975 30 103.9 5.29 1091.77 Chain B, Human Complement Component C3b gi110590600 52.22 903 29 74.8 7.12 1859.56 Chain B, Apo-Human Serum Transferrin (Glycosylated) gi317455058 64.17 761 29 71.0 7.30 891.66 Chain C, Crystal Structure Of Complement C3b In Complex With Factors B And D gi157831597 38.81 582 28 120.0 5.68 888.21 Chain A, X-Ray Crystal Structure Of Human Ceruloplasmin At 3.0 Angstroms gi81175167 27.58 334 26 192.7 7.15 517.84 Complement C4-B gi178557739 26.89 339 25 192.6 7.27 506.65 complement C4-B preproprotein -

Clinical Study Molecular Characterization of Schizophrenia Viewed by Microarray Analysis of Gene Expression in Prefrontal Cortex
Neuron, Vol. 28, 53±67, October, 2000, Copyright 2000 by Cell Press Molecular Characterization of Clinical Study Schizophrenia Viewed by Microarray Analysis of Gene Expression in Prefrontal Cortex Ka roly Mirnics,*³§ Frank A. Middleton,* pus, superior temporal gyrus, and thalamus, appear to Adriana Marquez,* David A. Lewis,² be disturbed in this disorder (Harrison, 1999; McCarley and Pat Levitt*³ et al., 1999). In particular, a convergence of observations *Department of Neurobiology from clinical, neuroimaging, and postmortem studies ² Departments of Psychiatry and Neuroscience have implicated the dorsal prefrontal cortex (PFC) as a ³ PittArray major locus of dysfunction in schizophrenia (Weinberger University of Pittsburgh School of Medicine et al., 1986; Selemon et al., 1995; Andreasen et al., 1997; Pittsburgh, Pennsylvania 15261 Bertolino et al., 2000). Abnormal PFC function probably contributes to many of the cognitive disturbances in schizophrenia and appears to be related to altered syn- aptic structure and/or function in this cortical region. Summary For example, in subjects with schizophrenia, reductions in gray matter volume in the dorsal PFC have been ob- Microarray expression profiling of prefrontal cortex served in neuroimaging studies (Goldstein et al., 1999; from matched pairs of schizophrenic and control sub- Sanfilipo et al., 2000), and these volumetric changes jects and hierarchical data analysis revealed that tran- are associated with an increase in cell packing density scripts encoding proteins involved in the regulation (Selemon et al., 1995, 1998; Lewis and Lieberman, 2000) of presynaptic function (PSYN) were decreased in all but no change in total neuron number in the PFC (Pak- subjects with schizophrenia. Genes of the PSYN group kenberg, 1993). -

Complete Genome of the Cellulolytic Thermophile Acidothermus Cellulolyticus 11B Provides Insights Into Its Ecophysiological and Evolutionary Adaptations
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus 11B provides insights into its ecophysiological and evolutionary adaptations Ravi D. Barabote,1,9 Gary Xie,1 David H. Leu,2 Philippe Normand,3 Anamaria Necsulea,4 Vincent Daubin,4 Claudine Me´digue,5 William S. Adney,6 Xin Clare Xu,2 Alla Lapidus,7 Rebecca E. Parales,8 Chris Detter,1 Petar Pujic,3 David Bruce,1 Celine Lavire,3 Jean F. Challacombe,1 Thomas S. Brettin,1 and Alison M. Berry2,10 1DOE Joint Genome Institute, Bioscience Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA; 2Department of Plant Sciences, University of California, Davis, California 95616, USA; 3Centre National de la Recherche Scientifique (CNRS), UMR5557, E´cologie Microbienne, Universite´ Lyon I, Villeurbanne F-69622, France; 4Centre National de la Recherche Scientifique (CNRS), UMR5558, Laboratoire de Biome´trie et Biologie E´volutive, Universite´ Lyon I, Villeurbanne F-69622, France; 5Centre National de la Recherche Scientifique (CNRS), UMR8030 and CEA/DSV/IG/Genoscope, Laboratoire de Ge´nomique Comparative, 91057 Evry Cedex, France; 6National Renewable Energy Laboratory, Golden, Colorado 80401, USA; 7DOE Joint Genome Institute, Walnut Creek, California 94598, USA; 8Department of Microbiology, University of California, Davis, California 95616, USA We present here the complete 2.4-Mb genome of the cellulolytic actinobacterial thermophile Acidothermus cellulolyticus 11B. New secreted glycoside hydrolases and carbohydrate esterases were identified in the genome, revealing a diverse biomass- degrading enzyme repertoire far greater than previously characterized and elevating the industrial value of this organism. -

Frontiersin.Org 1 April 2015 | Volume 9 | Article 123 Saunders Et Al
ORIGINAL RESEARCH published: 28 April 2015 doi: 10.3389/fnins.2015.00123 Influx mechanisms in the embryonic and adult rat choroid plexus: a transcriptome study Norman R. Saunders 1*, Katarzyna M. Dziegielewska 1, Kjeld Møllgård 2, Mark D. Habgood 1, Matthew J. Wakefield 3, Helen Lindsay 4, Nathalie Stratzielle 5, Jean-Francois Ghersi-Egea 5 and Shane A. Liddelow 1, 6 1 Department of Pharmacology and Therapeutics, University of Melbourne, Parkville, VIC, Australia, 2 Department of Cellular and Molecular Medicine, University of Copenhagen, Copenhagen, Denmark, 3 Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia, 4 Institute of Molecular Life Sciences, University of Zurich, Zurich, Switzerland, 5 Lyon Neuroscience Research Center, INSERM U1028, Centre National de la Recherche Scientifique UMR5292, Université Lyon 1, Lyon, France, 6 Department of Neurobiology, Stanford University, Stanford, CA, USA The transcriptome of embryonic and adult rat lateral ventricular choroid plexus, using a combination of RNA-Sequencing and microarray data, was analyzed by functional groups of influx transporters, particularly solute carrier (SLC) transporters. RNA-Seq Edited by: Joana A. Palha, was performed at embryonic day (E) 15 and adult with additional data obtained at University of Minho, Portugal intermediate ages from microarray analysis. The largest represented functional group Reviewed by: in the embryo was amino acid transporters (twelve) with expression levels 2–98 times Fernanda Marques, University of Minho, Portugal greater than in the adult. In contrast, in the adult only six amino acid transporters Hanspeter Herzel, were up-regulated compared to the embryo and at more modest enrichment levels Humboldt University, Germany (<5-fold enrichment above E15).