To Divide Or Differentiate? Nestin-Mediated Regulation of Cdk5 in Cell Fate Decisions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Genome-Wide Library of MADM Mice for Single-Cell Genetic Mosaic Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.05.136192; this version posted June 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Contreras et al., A Genome-wide Library of MADM Mice for Single-Cell Genetic Mosaic Analysis Ximena Contreras1, Amarbayasgalan Davaatseren1, Nicole Amberg1, Andi H. Hansen1, Johanna Sonntag1, Lill Andersen2, Tina Bernthaler2, Anna Heger1, Randy Johnson3, Lindsay A. Schwarz4,5, Liqun Luo4, Thomas Rülicke2 & Simon Hippenmeyer1,6,# 1 Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria 2 Institute of Laboratory Animal Science, University of Veterinary Medicine Vienna, Vienna, Austria 3 Department of Biochemistry and Molecular Biology, University of Texas, Houston, TX 77030, USA 4 HHMI and Department of Biology, Stanford University, Stanford, CA 94305, USA 5 Present address: St. Jude Children’s Research Hospital, Memphis, TN 38105, USA 6 Lead contact #Correspondence and requests for materials should be addressed to S.H. ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.05.136192; this version posted June 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Contreras et al., SUMMARY Mosaic Analysis with Double Markers (MADM) offers a unique approach to visualize and concomitantly manipulate genetically-defined cells in mice with single-cell resolution. -

Genetic Mosaic Dissection of Lis1 and Ndel1 in Neuronal Migration
Neuron Article Genetic Mosaic Dissection of Lis1 and Ndel1 in Neuronal Migration Simon Hippenmeyer,1,* Yong Ha Youn,2 Hyang Mi Moon,2,3 Kazunari Miyamichi,1 Hui Zong,1,5 Anthony Wynshaw-Boris,2,4 and Liqun Luo1,* 1Howard Hughes Medical Institute and Department of Biology, Stanford University, Stanford, CA 94305, USA 2Department of Pediatrics and Institute for Human Genetics 3Biomedical Sciences Graduate Program 4Eli and Edythe Broad Center of Regeneration Medicine and Stem Cell Research University of California, San Francisco School of Medicine, San Francisco, CA 94143, USA 5Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA *Correspondence: [email protected] (S.H.), [email protected] (L.L.) DOI 10.1016/j.neuron.2010.09.027 SUMMARY studied. Cortical layering occurs in an ‘‘inside-out’’ fashion whereby earlier born neurons occupy deep layers and succes- Coordinated migration of newly born neurons to their sively later born neurons settle in progressively upper layers (An- prospective target laminae is a prerequisite for neural gevine and Sidman, 1961; Rakic, 1974). Upon radial glia progen- circuit assembly in the developing brain. The evolu- itor cell (RGPC)-mediated neurogenesis, newborn migrating tionarily conserved LIS1/NDEL1 complex is essential cortical projection neurons are bipolar-shaped in the ventricular for neuronal migration in the mammalian cerebral zone (VZ) but then convert to a multipolar morphology within the cortex. The cytoplasmic nature of LIS1 and NDEL1 subventricular zone (SVZ) and migrate into the intermediate zone (IZ). A switch from the multipolar state back to a bipolar proteins suggest that they regulate neuronal migra- morphology precedes radial glia-guided locomotion of projec- tion cell autonomously. -

Activation of Aurora-A Is Essential for Neuronal Migration Via Modulation of Microtubule Organization
11050 • The Journal of Neuroscience, August 8, 2012 • 32(32):11050–11066 Cellular/Molecular Activation of Aurora-A Is Essential for Neuronal Migration via Modulation of Microtubule Organization Takako Takitoh,1 Kanako Kumamoto,1 Chen-Chi Wang,2,4 Makoto Sato,2,3,4 Shiori Toba,1 Anthony Wynshaw-Boris,5 and Shinji Hirotsune1 1Department of Genetic Disease Research, Osaka City University Graduate School of Medicine, Osaka 545-8585, Japan, 2Division of Cell Biology and Neuroscience, Department of Morphological and Physiological Sciences, Faculty of Medical Sciences, University of Fukui, 3Child Development Research Center, Graduate School of Medical Sciences, University of Fukui, 4Research and Education Program for Life Science, University of Fukui, Fukui 910-1193, Japan, and 5Department of Pediatrics and Institute for Human Genetics, University of California, San Francisco, School of Medicine, San Francisco, California 94143 Neuronal migration is a critical feature to ensure proper location and wiring of neurons during cortical development. Postmitotic neurons migrate from the ventricular zone into the cortical plate to establish neuronal lamina in an “inside-out” gradient of maturation. Here, we report that the mitotic kinase Aurora-A is critical for the regulation of microtubule organization during neuronal migration via an Aurora-A–NDEL1 pathway in the mouse. Suppression of Aurora-A activity by inhibitors or siRNA resulted in severe impairment of neuronal migration of granular neurons. In addition, in utero injection of the Aurora-A kinase-dead mutant provoked defective migra- tion of cortical neurons. Furthermore, we demonstrated that suppression of Aurora-A impaired microtubule modulation in migrating neurons. Interestingly, suppression of CDK5 by an inhibitor or siRNA reduced Aurora-A activity and NDEL1 phosphorylation by Aurora-A, which led to defective neuronal migration. -

Forschungsbericht Science Report
FORSCHUNGSBERICHT SCIENCE REPORT2017/2018 ST. ANNA KINDERKREBSFORSCHUNG ST. ANNA CHILDREN’S CANCER RESEARCH INSTITUTE TUMOR BIOLOGY Understanding tumor heterogeneity and relapse in neuroblastoma patients to allow adequate treatment options. MOLECULAR MICROBIOLOGY LCH BIOLOGY Clinically important Targeted resistant mutations in Philadelphia inhibition chromosome-positive leukemias. of the MAPK pathway leads to a STUDIES & STATISTICS rapid and sustained clinical response in severe multisystem LCH. Busulfan and melphalan is considered standard high-dose chemotherapy for high-risk neuroblastoma. GENETICS OF LEUKEMIAS A novel genetic MOLECULAR BIOLOGY OF SOLID TUMORS subtype of leukemia. Understanding mechanisms EPIGENOME-BASED PRECISION MEDICINE of tumor cell plasticity EPIGENETIC and its role in diversity in Ewing sarcoma metastasis. Ewing sarcoma. MORE SCIENCE REPORTS INSIDE >>> [ 06 ] Forschungsbericht St. Anna Kinderkrebsforschung | Science Report St. Anna Children’s Cancer Research Institute Inhalt Einleitung Vorwort des Institutsleiters 10 Vorwort des wissenschaftlichen Direktors 12 30 Jahre – unseren Spendern sei Dank! 14 Daten & Fakten Kompetitive Drittmittel 2018 20 Zuweisung der Geldmittel 2018 20 Finanzierung 2018 20 Nationen 21 Personelle Zusammensetzung 21 PatientInnenaufkommen im S2IRP 22 2-Jahres-Überlebensrate krebskranker Kinder 23 5-Jahres-Überlebensrate krebskranker Kinder 24 Forschungsnetzwerke 26 Inhalt Forschungsbericht I St. Anna Kinderkrebsforschung | Science Report St. Anna Cancer Research Institute [ 07 ] Science -

Lis1 and Ndel1 Influence the Timing of Nuclear Envelope Breakdown in Neural Stem Cells
University of South Carolina Scholar Commons Faculty Publications Biological Sciences, Department of 9-22-2008 Lis1 and Ndel1 Influence the Timing of Nuclear Envelope Breakdown in Neural Stem Cells Sachin Hebbar University of South Carolina - Columbia Mariano T. Mesngon University of South Carolina - Columbia Aimee M. Guillotte University of South Carolina - Columbia Bhavim Desai University of South Carolina - Columbia, [email protected] Ramses Ayala Mass General Institute for Neurodegenerative Disease See next page for additional authors Follow this and additional works at: https://scholarcommons.sc.edu/biol_facpub Part of the Biology Commons Publication Info The Journal of Cell Biology, Volume 182, Issue 6, 2008, pages 1063-1071. © 2008, the authors. Originally published in the Journal of Cell Biology by the Rockefeller University Press. This Article is brought to you by the Biological Sciences, Department of at Scholar Commons. It has been accepted for inclusion in Faculty Publications by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Author(s) Sachin Hebbar, Mariano T. Mesngon, Aimee M. Guillotte, Bhavim Desai, Ramses Ayala, and Deanna S. Smith This article is available at Scholar Commons: https://scholarcommons.sc.edu/biol_facpub/58 Published September 22, 2008 JCB: REPORT Lis1 and Ndel1 infl uence the timing of nuclear envelope breakdown in neural stem cells Sachin Hebbar , 1 Mariano T. Mesngon , 1 Aimee M. Guillotte , 1 Bhavim Desai , 1 Ramses Ayala , 2 and Deanna S. Smith 1 1 Department of Biological Sciences, University of South Carolina, Columbia, SC 29208 2 Neurology Department, Mass General Institute for Neurodegenerative Disease, Charlestown, MA 02120 is1 and Ndel1 are essential for animal development. -

Current Treatment of Juvenile Myelomonocytic Leukemia
Journal of Clinical Medicine Review Current Treatment of Juvenile Myelomonocytic Leukemia Christina Mayerhofer 1 , Charlotte M. Niemeyer 1,2 and Christian Flotho 1,2,* 1 Division of Pediatric Hematology and Oncology, Department of Pediatrics and Adolescent Medicine, Medical Center, Faculty of Medicine, University of Freiburg, 79106 Freiburg, Germany; [email protected] (C.M.); [email protected] (C.M.N.) 2 German Cancer Consortium (DKTK), 79106 Freiburg, Germany * Correspondence: christian.fl[email protected] Abstract: Juvenile myelomonocytic leukemia (JMML) is a rare pediatric leukemia characterized by mutations in five canonical RAS pathway genes. The diagnosis is made by typical clinical and hematological findings associated with a compatible mutation. Although this is sufficient for clinical decision-making in most JMML cases, more in-depth analysis can include DNA methylation class and panel sequencing analysis for secondary mutations. NRAS-initiated JMML is heterogeneous and adequate management ranges from watchful waiting to allogeneic hematopoietic stem cell transplan- tation (HSCT). Upfront azacitidine in KRAS patients can achieve long-term remissions without HSCT; if HSCT is required, a less toxic preparative regimen is recommended. Germline CBL patients often experience spontaneous resolution of the leukemia or exhibit stable mixed chimerism after HSCT. JMML driven by PTPN11 or NF1 is often rapidly progressive, requires swift HSCT and may benefit from pretransplant therapy with azacitidine. Because graft-versus-leukemia alloimmunity is central to cure high risk patients, the immunosuppressive regimen should be discontinued early after HSCT. Keywords: juvenile myelomonocytic leukemia; RAS signaling; hematopoietic stem cell transplantation; 5-azacitidine; myelodysplastic/myeloproliferative disorders; targeted therapy Citation: Mayerhofer, C.; Niemeyer, C.M.; Flotho, C. -

CRL5-Dependent Regulation of the Small Gtpases ARL4C and ARF6 Controls Hippocampal Morphogenesis
CRL5-dependent regulation of the small GTPases ARL4C and ARF6 controls hippocampal morphogenesis Jisoo S. Hana,1, Keiko Hinoa,1, Wenzhe Lia, Raenier V. Reyesa, Cesar P. Canalesa,2, Adam M. Miltnera, Yasmin Haddadia, Junqing Sunb, Chao-Yin Chenb, Anna La Torrea, and Sergi Simóa,3 aDepartment of Cell Biology and Human Anatomy, University of California, Davis, CA 95616; and bDepartment of Pharmacology, University of California, Davis, CA 95616 Edited by Mary E. Hatten, Rockefeller University, New York, NY, and approved August 3, 2020 (received for review February 14, 2020) The small GTPase ARL4C participates in the regulation of cell migra- Importantly, CRL5 substrate adaptors are dynamically regulated tion, cytoskeletal rearrangements, and vesicular trafficking in epithe- during development, directing CRL5 activity to specific substrates lial cells. The ARL4C signaling cascade starts by the recruitment of the at different times and areas in the CNS (19). In the neocortex, ARF–GEF cytohesins to the plasma membrane, which, in turn, bind CRL5 opposes projection neuron migration by down-regulating and activate the small GTPase ARF6. However, the role of ARL4C– the Reelin/DAB1 signaling pathway as migrating projection neu- cytohesin–ARF6 signaling during hippocampal development remains rons reach the top of the cortical plate (18, 20, 21). In the CNS, elusive. Here, we report that the E3 ubiquitin ligase Cullin 5/RBX2 Reelin/DAB1 signaling participates in several developmental (CRL5) controls the stability of ARL4C and its signaling effectors to processes, including neuron migration, dendritogenesis, axo- regulate hippocampal morphogenesis. Both RBX2 knockout and genesis, and synaptogenesis (22–24). Moreover, CRL5 also con- Cullin 5 knockdown cause hippocampal pyramidal neuron mislocali- trols levels of the tyrosine kinase FYN, which exerts important zation and development of multiple apical dendrites. -

Regulation of Titin-Based Cardiac Stiffness by Unfolded Domain Oxidation (Undox)
Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx) Christine M. Loeschera,1, Martin Breitkreuzb,1, Yong Lia, Alexander Nickelc, Andreas Ungera, Alexander Dietld, Andreas Schmidte, Belal A. Mohamedf, Sebastian Kötterg, Joachim P. Schmitth, Marcus Krügere,i, Martina Krügerg, Karl Toischerf, Christoph Maackc, Lars I. Leichertj, Nazha Hamdanib, and Wolfgang A. Linkea,2 aInstitute of Physiology II, University of Munster, 48149 Munster, Germany; bInstitute of Physiology, Ruhr University Bochum, 44801 Bochum, Germany; cComprehensive Heart Failure Center Wuerzburg, University Clinic Wuerzburg, 97078 Wuerzburg, Germany; dDepartment of Internal Medicine II, University Hospital Regensburg, 93053 Regensburg, Germany; eInstitute for Genetics, University of Cologne, 50931 Cologne, Germany; fDepartment of Cardiology and Pneumology, University Medicine Goettingen, 37075 Goettingen, Germany; gDepartment of Cardiovascular Physiology, Heinrich Heine University, 40225 Düsseldorf, Germany; hDepartment of Pharmacology and Clinical Pharmacology, Heinrich Heine University, 40225 Düsseldorf, Germany; iCenter for Molecular Medicine and Excellence Cluster "Cellular Stress Responses in Aging-Associated Diseases" (CECAD), University of Cologne, 50931 Cologne, Germany; and jInstitute for Biochemistry and Pathobiochemistry, Ruhr University Bochum, 44801 Bochum, Germany Edited by Jonathan Seidman, Harvard University, Boston, MA, and approved August 12, 2020 (received for review March 14, 2020) The relationship between oxidative stress and -

Postmortem Changes in the Myofibrillar and Other Cytoskeletal Proteins in Muscle
BIOCHEMISTRY - IMPACT ON MEAT TENDERNESS Postmortem Changes in the Myofibrillar and Other C'oskeletal Proteins in Muscle RICHARD M. ROBSON*, ELISABETH HUFF-LONERGAN', FREDERICK C. PARRISH, JR., CHIUNG-YING HO, MARVIN H. STROMER, TED W. HUIATT, ROBERT M. BELLIN and SUZANNE W. SERNETT introduction filaments (titin), and integral Z-line region (a-actinin, Cap Z), as well as proteins of the intermediate filaments (desmin, The cytoskeleton of "typical" vertebrate cells contains paranemin, and synemin), Z-line periphery (filamin) and three protein filament systems, namely the -7-nm diameter costameres underlying the cell membrane (filamin, actin-containing microfilaments, the -1 0-nm diameter in- dystrophin, talin, and vinculin) are listed along with an esti- termediate filaments (IFs), and the -23-nm diameter tubu- mate of their abundance, approximate molecular weights, lin-containing microtubules (Robson, 1989, 1995; Robson and number of subunits per molecule. Because the myofibrils et al., 1991 ).The contractile myofibrils, which are by far the are the overwhelming components of the skeletal muscle cell major components of developed skeletal muscle cells and cytoskeleton, the approximate percentages of the cytoskel- are responsible for most of the desirable qualities of muscle eton listed for the myofibrillar proteins (e.g., myosin, actin, foods (Robson et al., 1981,1984, 1991 1, can be considered tropomyosin, a-actinin, etc.) also would represent their ap- the highly expanded corollary of the microfilament system proximate percentages of total myofibrillar protein. of non-muscle cells. The myofibrils, IFs, cell membrane skel- eton (complex protein-lattice subjacent to the sarcolemma), Some Important Characteristics, Possible and attachment sites connecting these elements will be con- Roles, and Postmortem Changes of Key sidered as comprising the muscle cell cytoskeleton in this Cytoskeletal Proteins review. -

Α-Actinin/Titin Interaction: a Dynamic and Mechanically Stable Cluster of Bonds in the Muscle Z-Disk
α-Actinin/titin interaction: A dynamic and mechanically stable cluster of bonds in the muscle Z-disk Marco Grisona, Ulrich Merkela, Julius Kostanb, Kristina Djinovic-Carugob,c, and Matthias Riefa,d,1 aPhysik Department E22, Technische Universität München, 85748 Garching, Germany; bDepartment of Structural and Computational Biology, Max F. Perutz Laboratories, University of Vienna, A-1030 Vienna, Austria; cDepartment of Biochemistry, Faculty of Chemistry and Chemical Technology, University of Ljubljana, SI-1000 Ljubljana, Slovenia; and dMunich Center for Integrated Protein Science, 81377 Munich, Germany Edited by James A. Spudich, Stanford University School of Medicine, Stanford, CA, and approved December 16, 2016 (received for review August 2, 2016) Stable anchoring of titin within the muscle Z-disk is essential for In humans, the isoforms of titin exhibit four to seven Z-repeats preserving muscle integrity during passive stretching. One of the (15, 16, 20). The structure of the EF3-4 hands complex with titin main candidates for anchoring titin in the Z-disk is the actin cross- Z-repeat 7 shows the bound Z-repeat in an α-helical confor- linker α-actinin. The calmodulin-like domain of α-actinin binds to mation (21). In solution assays, binding affinities of various the Z-repeats of titin. However, the mechanical and kinetic prop- Z-repeats to EF3-4 were determined to lie in the micromolar erties of this important interaction are still unknown. Here, we use range (22). Micromolar affinity points only to a moderately a dual-beam optical tweezers assay to study the mechanics of this stable interaction, the kinetics of which are unknown. -
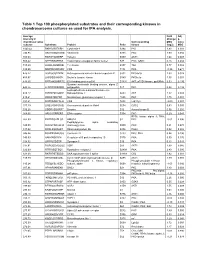
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Association of Titin and Myosin Heavy Chain in Developing Skeletal Muscle (Myogenesis/Cytoskeleton/Assembly in Vvo) W
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 74%-7500, August 1992 Cell Biology Association of titin and myosin heavy chain in developing skeletal muscle (myogenesis/cytoskeleton/assembly in vvo) W. B. ISAACS*, I. S. KIM, A. STRUVE, AND A. B. FULTONt Department of Biochemistry, University of Iowa, Iowa City, IA 52242 Communicated by Sheldon Penman, May 22, 1992 ABSTRACT To understand molecular interactions that deficient medium (10). After labeling, cultures either were organize developing myoflbrils, we examined the biosynthesis extracted immediately or were chased by adding complete and interaction of titin and myosin heavy chain in cultures of medium supplemented with 2 mM unlabeled methionine and developing muscle. Use of pulse-labeling, immunoprecipita- incubating at 370C for various times before extraction. Ex- tion, and a reversible cross-linking procedure demonstrates tractions used 0.5% Triton X-100 in extraction buffer (100 that within minutes of synthesis, titin and myosin heavy chain mM KCI/10 mM Pipes, pH 6.8/300 mM sucrose/2 mM can be chemically cross-linked into very large, detergent- MgCI2/1 mM EGTA) containing protease inhibitors (1 mM resistant complexes retaining many features of intact myo- phenylmethylsulfonyl fluoride and 100 mM leupeptin; ref. tubes. These complexes, predominantly of titin and myosin, 11). occur very early in myofibrillogenesis as well as later. These Immunoprecipitation. Titin was precipitated by using a data suggest that synthesis and assembly oftitin and myosin are mouse monoclonal antibody (mAb), AMF-1, as described temporally and spatially coordinated in nascent myofibrils and (10). Muscle-specific myosin heavy chain (hereafter myosin) support the hypothesis that titin molecules help to organize was precipitated with mAb MF-20 (12), a gift ofD.