Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Baseline Levels of Endogenous Compounds in the Caucasian Population S
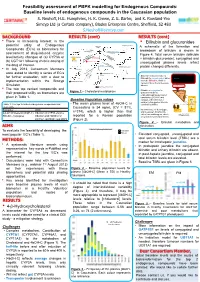
Feasibility assessment of PBPK modelling for Endogenous Compounds: Baseline levels of endogenous compounds in the Caucasian population S. Neuhoff, H.E. Humphries, H. K. Crewe, Z. E. Barter, and K. Rowland-Yeo Simcyp Ltd (a Certara company), Blades Enterprise Centre, Sheffield, S2 4SU [email protected] BACKGROUND RESULTS (cont) RESULTS (cont) • There is increasing interest in the • Bilirubin and glucuronides 27-Hydroxycholesterol potential utility of Endogenous Cholesterol 1g/day A schematic of the formation and Compounds (EC’s) as biomarkers for CYP7A1 (liver) 500 mg/day CYP27A1 (many tissues) breakdown of bilirubin is shown in assessment of drug-induced enzyme 19 mg/day Figure 4. Total serum bilirubin (bilirubin 7α-Hydroxycholesterol level/activity changes of (a) CYP3A or CYP3A4 + bilirubin glucuronide), conjugated and Bile Acids (b) UGT1A1 following chronic dosing of 4-Hydroxycholesterol unconjugated plasma levels reflect the drug of interest. CYP27A1 (liver and extrahepatic) 24S-Hydroxycholesterol protein changes differently. • In July 2013, Consortium Members Pregnenolone were asked to identify a series of EC’s 4,27-Dihydroxycholesterol Macrophage CYP27A1 • Biliverdin reductase reduces Heme Hemeoxygenase for further evaluation, with a view to 4,24-Dihydroxycholesterol Biliverdin to unconjugated, water- Biliverdin 4,7α-Dihydroxycholesterol insoluble Bilirubin, which is Biliverdin reductase implementation within the Simcyp Bilirubin carried in blood bound to serum Unconjugated bilirubin Simulator. albumin complexed with albumin 3 -

Based Photodynamic Diagnosis of Malignant Brain Tumor: Molecular Design of ABCG2 Inhibitors
Pharmaceutics 2011 , 3, 615-635; doi:10.3390/pharmaceutics3030615 OPEN ACCESS pharmaceutics ISSN 1999-4923 www.mdpi.com/journal/pharmaceutics Review Transporter-Mediated Drug Interaction Strategy for 5-Aminolevulinic Acid (ALA)-Based Photodynamic Diagnosis of Malignant Brain Tumor: Molecular Design of ABCG2 Inhibitors Toshihisa Ishikawa 1,*, Kenkichi Takahashi 2, Naokado Ikeda 2, Yoshinaga Kajimoto 2, Yuichiro Hagiya 3, Shun-ichiro Ogura 3, Shin-ichi Miyatake 2 and Toshihiko Kuroiwa 2 1 Omics Science Center, RIKEN Yokohama Institute, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, 230-0045, Japan 2 Department of Neurosurgery, Osaka Medical College, Osaka 569-8686, Japan 3 Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, Yokohama 226-8501, Japan * Author to whom correspondence should be addressed: E-Mail: [email protected]; Tel.: +81-45-503-9222; Fax: +81-45-503-9216. Received: 19 July 2011; in revised form: 16 August 2011 / Accepted: 9 September 2011 / Published: 14 September 2011 Abstract: Photodynamic diagnosis (PDD) is a practical tool currently used in surgical operation of aggressive brain tumors, such as glioblastoma. PDD is achieved by a photon-induced physicochemical reaction which is induced by excitation of protoporphyrin IX (PpIX) exposed to light. Fluorescence-guided gross-total resection has recently been developed in PDD, where 5-aminolevulinic acid (ALA) or its ester is administered as the precursor of PpIX. ALA induces the accumulation of PpIX, a natural photo-sensitizer, in cancer cells. Recent studies provide evidence that adenosine triphosphate (ATP)-binding cassette (ABC) transporter ABCG2 plays a pivotal role in regulating the cellular accumulation of porphyrins in cancer cells and thereby affects the efficacy of PDD. -

Bilirubin Secretion, Jaundice and Evaluation of Liver Function
Bilirubin Secretion, Jaundice and Evaluation of Liver Function Howard J. Worman, M. D. Evaluation of Liver Disease and Hepatic Function History Physical Examination Laboratory Tests Sometimes Radiological/Nuclear Medicine Sometimes Liver Biopsy 1 Jaundice occurs as a result of excess bilirubin in the blood. It is a hallmark of liver disease but not always present in liver disease. Jaundice occurs when the liver fails to adequately secrete bilirubin from the blood into the bile. To understand how jaundice occurs, you must first understand bilirubin synthesis, metabolism and secretion. 2 Heme Oxygenase Schuller et al. Nature Structural Biology 6, 860 - 867 (1999) Bilivirdin Reductase Kikuchi et al. Nature Structural Biology 8, 221 - 225 (2001) 3 Bilirubin is frequently depicted as a linear tetrapyrrole. However, intramolecular hydrogen bonding fixes it in a rigid structure that blocks exposure of its polar groups to aqueous solvents, making it very insoluble in blood. 4 Bilirubin in Blood is Bound to Albumin: Uptake into Hepatocyte at Basolateral (Sinusoidal) Membrane Some OATP-C? bilirubin stored in cytosol bound to proteins Bilurubin UDP-glucuronosyltransferase is localized to the endo- plasmic reticulum; it catalyzes conjugation to a diglucuronide, making it more water soluble. A: Labeling of periphery of cell hepatocyte nucleus B: Labeling of ER with antibody to UDP-glucuronosyltransferase 5 Alternative RNA splicing of different first exons of UGT1 gives different isoforms with different substrate specificities, some for bilirubin and others -

Inherited Disorders of Bilirubin Clearance N
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Hofstra Northwell Academic Works (Hofstra Northwell School of Medicine) Donald and Barbara Zucker School of Medicine Journal Articles Academic Works 2015 Inherited disorders of bilirubin clearance N. Memon B. I. Weinberger Zucker School of Medicine at Hofstra/Northwell T. Hegyi L. M. Aleksunes Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Pediatrics Commons Recommended Citation Memon N, Weinberger B, Hegyi T, Aleksunes L. Inherited disorders of bilirubin clearance. 2015 Jan 01; 79(3):Article 2776 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/2776. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. For more information, please contact [email protected]. HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Pediatr Manuscript Author Res. Author manuscript; Manuscript Author available in PMC 2016 April 05. Published in final edited form as: Pediatr Res. 2016 March ; 79(3): 378–386. doi:10.1038/pr.2015.247. Inherited Disorders of Bilirubin Clearance Naureen Memon1,*, Barry I Weinberger2, Thomas Hegyi1, and Lauren M Aleksunes3 1Department of Pediatrics, Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA 2Department of Pediatrics, Cohen Children’s Medical Center of New York, New Hyde Park, NY, USA 3Department of Pharmacology and Toxicology, Rutgers University, Piscataway, NJ, USA Abstract Inherited disorders of hyperbilirubinemia may be caused by increased bilirubin production or decreased bilirubin clearance. -

Novel Pharmacogenomic Markers of Irinotecan-Induced Severe Toxicity in Metastatic Colorectal Cancer Patients
Novel pharmacogenomic markers of irinotecan-induced severe toxicity in metastatic colorectal cancer patients Thèse Siwen Sylvialin Chen Doctorat en sciences pharmaceutiques Philosophiae doctor (Ph.D.) Québec, Canada © Siwen Sylvialin Chen, 2015 ii Résumé L’irinotécan est un agent de chimiothérapie largement utilisé pour le traitement de tumeurs solides, particulièrement pour le cancer colorectal métastatique (mCRC). Fréquemment, le traitement par l’irinotécan conduit à la neutropénie et la diarrhée, des effets secondaires sévères qui peuvent limiter la poursuite du traitement et la qualité de vie des patients. Plusieurs études pharmacogénomiques ont évalué les risques associés à la chimiothérapie à base d’irinotécan, en particulier en lien avec le gène UGT1A, alors que peu d’études ont examiné l’impact des gènes codant pour des transporteurs. Par exemple, le marqueur UGT1A1*28 a été associé à une augmentation de 2 fois du risque de neutropénie, mais ce marqueur ne permet pas de prédire la toxicité gastrointestinale ou l’issue clinique. L’objectif de cette étude était de découvrir de nouveaux marqueurs génétiques associés au risque de toxicité induite par l’irinotécan, en utilisant une stratégie d’haplotype/SNP-étiquette permettant de maximiser la couverture des loci génétiques ciblés. Nous avons analysé les associations génétiques des loci UGT1 et sept gènes codants pour des transporteurs ABC impliqués dans la pharmacocinétique de l’irinotécan, soient ABCB1, ABCC1, ABCC2, ABCC5, ABCG1, ABCG2 ainsi que SLCO1B1. Les profils de 167 patients canadiens atteints de mCRC sous traitement FOLFIRI (à base d’irinotécan) ont été examinés et les marqueurs significatifs ont par la suite été validés dans une cohorte indépendante de 250 patients italiens. -

Mechanism of Bilirubin Diglucuronide Formation in Intact Rats: BILIRUBIN
Mechanism of Bilirubin Diglucuronide Formation in Intact Rats BILIRUBIN DIGLUCURONIDE FORMATION IN VIVO NORBERT BLANCKAERT, JOHN GOLLAN, and RUDI SCHMID, Department of Medicine and Liver Center, University of California School of Medicine, San Francisco, California 94143; Laboratory of Hepatology, Department of Medical Research, University of Leuven, B-3000, Leuven, Belgium A B S T R A C T Although it is well established that bili- ['4C]monoglucuronide, no transfer or exchange of the rubin monoglucuronide is formed in the liver from bili- ['4C]glucuronosyl group between injected and en- rubin by a microsomal bilirubin uridine diphosphate dogenously produced bilirubin monoglucuronide (UDP)-glucuronosyltransferase, the subcellular site of could be detected in the excreted bilirubin diglucuron- conversion of monoglucuronide to diglucuronide and ide. Third, in homozygous Gunn rats, injected "4C- the molecular mechanism involved in diglucuronide labeled or unlabeled bilirubin mono- or digluc- synthesis have not been identified. Based on in vitro uronides were excreted in bile unchanged (except that studies, it has been proposed that two fundamentally diglucuronide was hydrolyzed to a minor degree). This different enzyme systems may be involved in di- indicates that Gunn rats, which lack bilirubin UDP- glucuronide synthesis in rat liver: (a) a microsomal glucuronosyltransferase activity, are unable to convert UDP-glucuronosyltransferase system requiring UDP- injected monoglucuronide to diglucuronide. Collec- glucuronic acid as sugar donor or (b) a transglucuroni- tively, these findings establish that a transglucuronida- dation mechanism that involves transfer of a glucurono- tion mechanism is not operational in vivo and support syl residue from one monoglucuronide molecule to the concept that bilirubin diglucuronide is formed by another, catalyzed by a liver plasma membrane en- a microsomal UDP-glucuronosyltransferase system. -

International Symposium on Bile Pigments
Gut: first published as 10.1136/gut.7.6.706 on 1 December 1966. Downloaded from Gut, 1966, 7, 706 International symposium on bile pigments An international symposium on bile pigments was 100 ml. In normal human adults (blood bank doners) the held at the Royal Free Hospital, London, on 11 and serum bilirubin glucuronide concentrations ranged from 12 July 1966. 5 to 25 ,g. per 100 ml. Male donors had significantly higher serum levels than females. ORIGIN OF BILE PIGMENTS B. NOIR (Buenos Aires) presented chromatographic evi- L G. ISRAELS (Winnipeg) produced evidence that the dence which showed that 10 to 15 % of the conjugated bile early labelled fraction of bilirubin which could be demon- pigments was present as bilirubin sulphate. strated using isotopically labelled glycine was composed of two components, one related to erythropoiesis and the E. TALAFANT (Czechoslovakia) studied the nature of other apparently unrelated to erythropoiesis and probably ether-extractable bilirubin in obstructive jaundice caused of hepatic origin. by carcinoma. He postulated that this was a complex of bilirubin glucuronide with an unknown lipid substance. S. SCHWARTZ (Minneapolis) emphasized that the early labelled bilirubin fraction probably had many compon- J. D. OSTROW (Cleveland) examined the yellow pigments ents each with its own turnover rate and that this made in bile of the Gunn rat and showed that they could not be quantitation ofeach component all the more difficult. identified with the yellow pigments formed from the photooxidative breakdown of bilirubin. S. H. ROBINSON (Boston) described a new experimental technique for studying the early labelled component of M. -

Molecular Genetics of Variegate Porphyria in Finland
Department of Medicine, Division of Endocrinology University of Helsinki, Finland MOLECULAR GENETICS OF VARIEGATE PORPHYRIA IN FINLAND Mikael von und zu Fraunberg Academic dissertation To be presented with the permission of the Medical Faculty of the University of Helsinki, for public examination in Auditorium II, Haartmaninkatu 8, Biomedicum Helsinki, on March 22th 2003, at 12:00 noon Helsinki 2003 SUPERVISED BY Docent Raili Kauppinen, M.D., Ph.D. Department of Medicine Division of Endocrinology University of Helsinki Finland REVIEWED BY Professor Richard J. Hift, M.D., Ph.D. Department of Medicine Lennox Eales Porphyria Laboratory MRC/UCT Liver Research Centre University of Cape Town South Africa and Professor Eeva-Riitta Savolainen, M.D., Ph.D. Department of Clinical Chemistry University of Oulu Finland OFFICIAL OPPONENT: Docent Katriina Aalto-Setälä, M.D., Ph.D. Department of Medicine University of Tampere Finland © Mikael von und zu Fraunberg ISBN 952-91-5643-X (paperback) ISBN 952-10-0970-5 (pdf) http://ethesis.helsinki.fi Helsinki 2003 Yliopistopaino to my family CONTENTS SUMMARY............................................................................................................... 6 ORIGINAL PUBLICATIONS................................................................................... 8 ABBREVIATIONS.................................................................................................... 9 INTRODUCTION ................................................................................................... 10 REVIEW -

7 Bilirubin Metabolism
#7 Bilirubin metabolism Objectives : ● Definition of bilirubin ● The normal plasma concentration of total bilirubin ● Bilirubin metabolism : - Bilirubin formation - Transport of bilirubin in plasma - Hepatic bilirubin transport - Excretion through intestine ● Other substances conjugated by glucuronyl transferase. ● Differentiation between conjugated & unconjugated bilirubin ● Other substances excreted in the bile ● Definition of Jaundice ● Classification of jaundice ( Prehepatic / Hepatic / poat-hepatic ). Doctors’ notes Extra Important Resources: 435 Boys’ & Girls’ slides | Guyton and Hall 12th & 13th edition Editing file [email protected] 1 Overview- mind map Porphyrin Metabolism (Boys’ slides) : ● Porphyrins are cyclic compounds that readily bind metal ions usually Fe2+ or +3 Fe which can carry O2. ● Porphyrins are heterocyclic macrocycles composed of four modified pyrrole (a colorless, toxic, liquid, five-membered ring compound, C4 H5 N) subunits interconnected at their α carbon atoms via methine bridges (=CH-). ● The most prevalent porphyrin in the human is heme, which consists of one ferrous (Fe2+ ) iron ion coordinated in the center of tetrapyrrole ring of protoporphyrin IX. ● Structure of Hemoglobin showing the polypeptides backbone that are composed of four subunits: 2 α and 2 β subunits. Every subunit is consisted of one ferrous (Fe2+ ) iron ion coordinated in the center porphyrin compound. The most prevalent porphyrin in the human is heme Definition of bilirubin : ● Bilirubin is the end product of heme degradation derived from breakdown senescent (aging) erythrocytes by mononuclear phagocytes system specially in the spleen, liver and bone marrow. (It is the water insoluble breakdown product of normal heme catabolism). ● Bilirubin is the greenish yellow pigment excreted in bile, urine and feces. ● The major pigment present in bile is the orange compound bilirubin. -

The Journal of the International Federation of Clinical Chemistry and Laboratory Medicine in This Issue
October 2019 ISSN 1650-3414 Volume 30 Number 3 Communications and Publications Division (CPD) of the IFCC Editor-in-chief: Prof. János Kappelmayer, MD, PhD Faculty of Medicine, University of Debrecen, Hungary e-mail: [email protected] The Journal of the International Federation of Clinical Chemistry and Laboratory Medicine In this issue Recommendations on measurement units – why and how Young Bae Lee Hansen 250 Evaluation of visual serum indices measurements and potential false result risks in routine clinical chemistry tests in Addis Ababa, Ethiopia Tigist Getahun, Anberber Alemu, Firehiwot Mulugeta, Merone Sileshi, Abenezer Ayalkebet, Wosene Habtu, Zeleke Geto, Fitsum Girma, Feyissa Challa, Mistire Wolde 276 Best practices in the implementation of a point of care testing program: experience from a tertiary care hospital in a developing country Aysha Habib Khan, Shahid Shakeel, Khairunnissa Hooda, Kashif Siddiqui, Lena Jafri 288 Prevalence of liver function test abnormality and associated factors in type 2 diabetes mellitus: a comparative cross-sectional study Getnet Teshome, Sintayehu Ambachew, Alebachew Fasil, Molla Abebe 303 Correlation of body mass index and waist/hip ratio with glycated hemoglobin in prediabetes Manju Bala, Meenakshi, Sameer Aggarwal 317 Hyperuricemia and its association with cardiovascular disease risk factors in type two diabetes mellitus patients at the University of Gondar Hospital, Northwest Ethiopia Birhanu Woldeamlak, Ketsela Yirdaw, Belete Biadgo 325 Atypical hemolytic uremic syndrome: genetic landscape challenge Laura Valiña, Bernardo López Andrade, Josep Miquel Bauça 340 Letter: Manchineel apple of death Michelle Muscat 346 This is a Platinum Open Access Journal distributed under the terms of the Creative Commons Attribution Non-Commercial License which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Xenobiotic regulation of Phase I and Phase II metabolism enzymes : beyond the Ah receptor paradigm Permalink https://escholarship.org/uc/item/5nz70459 Author Bonzo, Jessica A. Publication Date 2007 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Xenobiotic Regulation of Phase I and Phase II Metabolism Enzymes: Beyond the Ah Receptor Paradigm A Dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Jessica A. Bonzo Committee in charge: Professor Robert H. Tukey, Chair Professor Daniel J. Donoghue Professor Ronald M. Evans Professor Michael Karin Professor Julian I. Schroeder 2007 Copyright Jessica A. Bonzo, 2007 All rights reserved. The Dissertation of Jessica A. Bonzo is approved, and it is acceptable in quality and form for publication on microfilm: Chair University of California, San Diego 2007 iii Dedication To my parents, Roy T. and Flora E. A. Bonzo. iv Table of Contents Signature Page …………………………………………………. iii Dedication ……….……………………………………………... iv Table of Contents …………………………………………….... v List of Abbreviations .………………………………………...... vii List of Figures ………………..………………………………… xi List of Tables ............................................................................... xiv Acknowledgements ..................................................................... xv Vita ............................................................................................. -

“Cumulome” Reveals Major Metabolic Aberrations After Maturation in Vitro
Walter et al. BMC Genomics (2019) 20:588 https://doi.org/10.1186/s12864-019-5836-5 RESEARCHARTICLE Open Access Analysis of the equine “cumulome” reveals major metabolic aberrations after maturation in vitro Jasmin Walter1* , Fabian Huwiler1, Claudia Fortes2, Jonas Grossmann2, Bernd Roschitzki2, Junmin Hu2, Hanspeter Naegeli3, Endre Laczko2 and Ulrich Bleul1 Abstract Background: Maturation of oocytes under in vitro conditions (IVM) results in impaired developmental competence compared to oocytes matured in vivo. As oocytes are closely coupled to their cumulus complex, elucidating aberrations in cumulus metabolism in vitro is important to bridge the gap towards more physiological maturation conditions. The aim of this study was to analyze the equine “cumulome” in a novel combination of proteomic (nano-HPLC MS/MS) and metabolomic (UPLC-nanoESI-MS) profiling of single cumulus complexes of metaphase II oocytes matured either in vivo (n = 8) or in vitro (n = 7). Results: A total of 1811 quantifiable proteins and 906 metabolic compounds were identified. The proteome contained 216 differentially expressed proteins (p ≤ 0.05; FC ≥ 2; 95 decreased and 121 increased in vitro), and the metabolome contained 108 metabolites with significantly different abundance (p ≤ 0.05; FC ≥ 2; 24 decreased and 84 increased in vitro). The in vitro “cumulome” was summarized in the following 10 metabolic groups (containing 78 proteins and 21 metabolites): (1) oxygen supply, (2) glucose metabolism, (3) fatty acid metabolism, (4) oxidative phosphorylation, (5) amino acid metabolism, (6) purine and pyrimidine metabolism, (7) steroid metabolism, (8) extracellular matrix, (9) complement cascade and (10) coagulation cascade. The KEGG pathway “complement and coagulation cascades” (ID4610; n = 21) was significantly overrepresented after in vitro maturation.