Molecular Genetics of Variegate Porphyria in Finland
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical and Biochemical Characteristics and Genotype – Phenotype Correlation in Finnish Variegate Porphyria Patients
European Journal of Human Genetics (2002) 10, 649 – 657 ª 2002 Nature Publishing Group All rights reserved 1018 – 4813/02 $25.00 www.nature.com/ejhg ARTICLE Clinical and biochemical characteristics and genotype – phenotype correlation in Finnish variegate porphyria patients Mikael von und zu Fraunberg*,1, Kaisa Timonen2, Pertti Mustajoki1 and Raili Kauppinen1 1Department of Medicine, Division of Endocrinology, University Central Hospital of Helsinki, Biomedicum Helsinki, Helsinki, Finland; 2Department of Dermatology, University Central Hospital of Helsinki, Biomedicum Helsinki, Helsinki, Finland Variegate porphyria (VP) is an inherited metabolic disease resulting from the partial deficiency of protoporphyrinogen oxidase, the penultimate enzyme in the heme biosynthetic pathway. We have evaluated the clinical and biochemical outcome of 103 Finnish VP patients diagnosed between 1966 and 2001. Fifty-two per cent of patients had experienced clinical symptoms: 40% had photosensitivity, 27% acute attacks and 14% both manifestations. The proportion of patients with acute attacks has decreased dramatically from 38 to 14% in patients diagnosed before and after 1980, whereas the prevalence of skin symptoms had decreased only subtly from 45 to 34%. We have studied the correlation between PPOX genotype and clinical outcome of 90 patients with the three most common Finnish mutations I12T, R152C and 338G?C. The patients with the I12T mutation experienced no photosensitivity and acute attacks were rare (8%). Therefore, the occurrence of photosensitivity was lower in the I12T group compared to the R152C group (P=0.001), whereas no significant differences between the R152C and 338G?C groups could be observed. Biochemical abnormalities were significantly milder suggesting a milder form of the disease in patients with the I12T mutation. -

Liver Transplantation for the Management of Acute Intermittent
nd Gas y a tro g in lo t o e t s a t i p n Journal of Hepatology and e a l H D f i o s Kappus at al., J Hepatol Gastroint Dis 2017, 3:1 l o a r d ISSN:n 2475-3181 r e u r s o J Gastrointestinal Disorders DOI: 10.4172/2475-3181.1000141 Case Report Open Access Liver Transplantation for the Management of Acute Intermittent Porphyria: A Case Report Matthew R Kappus1, Bryant B Summers2, Jennifer S Byrns2*, Carl L Berg1 and Julius M Wilder1 1Division of Gastroenterology, Duke University School of Medicine, Durham NC, United States 2Department of Pharmacy, Duke University Hospital, Durham NC, United States *Corresponding author: Jennifer S Byrns, Department of Pharmacy, Duke University Hospital, DUMC BOX 3089, Durham, NC 27710, United States, Tel: 919-681-0677; E-mail: [email protected] Received date: December 27, 2016; Accepted date: January 16, 2017; Published date: January 20, 2017 Copyright: © 2017 Kappus MR, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract We report a single patient with acute intermittent porphyria whose porphyria was successfully treated with orthotopic liver transplantation. She had a very poor quality of life as a result of years of frequent acute porphyria symptoms manifesting as abdominal pain crises. After transplantation, clinical signs of porphyria resolved as expected. This case adds to a growing body of literature which is assisting to formulate the indications for, and the timing of, transplantation for AIP. -

Hereditary Corpoporphyria Presenting with Deep Jaundice and Photosensitivity
ANNALS OF GASTROENTEROLOGY 2001, 14(4):319-324 Case report Hereditary corpoporphyria presenting with deep jaundice and photosensitivity D. Kapetanos, P. Xiarhos, E. Kapetis1, A. Avgerinos, A. Ilias, G. Kokozidis, G. Kitis CASE REPORT SUMMARY A 20-year old woman was referred to our department Hereditary coproporphyria is a rare hepatic porphyria with from another hospital in May 2000, due to painless jaun- symptoms similar to acute intermittent porphyria. Photo- dice and pruritus. She reported having dark urine and sensitivity is not described in the latter. We report the case pale stools for two weeks and jaundice for 10 days be- of a young female with deep jaundice, photosensitivity, ane- fore admission. mia, renal failure and no abdominal pain. Hereditary co- proporphyria was diagnosed and the patient was discharged The patient had no past medical history, with no ne- after 111 days in full recovery. She was readmitted after 45 onatal jaundice, had received no medications during the days with abdominal pain and no jaundice or photosensi- previous months and had no alcohol consumption in the tivity. Severe neuropathy developed which caused tetrapare- past. She was on a diet in order to lose weight (her initial sis and deterioration of the respiratory muscle function. weight was 80 kg) and had been using home insecticides Haem arginate was administered with gradual improvement several days before. Her parents reported a family histo- of neuropathy. ry of thalassemia major. Key words: Hereditary corpoporphyria, jaundice, photosen- On admission she was icteric. She had 2-3 blisters on sitivity, neuropathy, haem arginate her face with serous yellow colored fluid. -
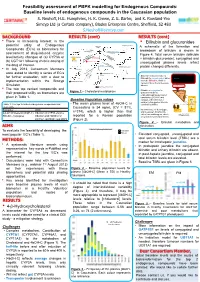
Baseline Levels of Endogenous Compounds in the Caucasian Population S
Feasibility assessment of PBPK modelling for Endogenous Compounds: Baseline levels of endogenous compounds in the Caucasian population S. Neuhoff, H.E. Humphries, H. K. Crewe, Z. E. Barter, and K. Rowland-Yeo Simcyp Ltd (a Certara company), Blades Enterprise Centre, Sheffield, S2 4SU [email protected] BACKGROUND RESULTS (cont) RESULTS (cont) • There is increasing interest in the • Bilirubin and glucuronides 27-Hydroxycholesterol potential utility of Endogenous Cholesterol 1g/day A schematic of the formation and Compounds (EC’s) as biomarkers for CYP7A1 (liver) 500 mg/day CYP27A1 (many tissues) breakdown of bilirubin is shown in assessment of drug-induced enzyme 19 mg/day Figure 4. Total serum bilirubin (bilirubin 7α-Hydroxycholesterol level/activity changes of (a) CYP3A or CYP3A4 + bilirubin glucuronide), conjugated and Bile Acids (b) UGT1A1 following chronic dosing of 4-Hydroxycholesterol unconjugated plasma levels reflect the drug of interest. CYP27A1 (liver and extrahepatic) 24S-Hydroxycholesterol protein changes differently. • In July 2013, Consortium Members Pregnenolone were asked to identify a series of EC’s 4,27-Dihydroxycholesterol Macrophage CYP27A1 • Biliverdin reductase reduces Heme Hemeoxygenase for further evaluation, with a view to 4,24-Dihydroxycholesterol Biliverdin to unconjugated, water- Biliverdin 4,7α-Dihydroxycholesterol insoluble Bilirubin, which is Biliverdin reductase implementation within the Simcyp Bilirubin carried in blood bound to serum Unconjugated bilirubin Simulator. albumin complexed with albumin 3 -

Ex Vivo Gene Therapy: a “Cultured” Surgical Approach to Curing Inherited Liver Disease
Mini Review Open Access J Surg Volume 10 Issue 3 - March 2019 Copyright © All rights are reserved by Joseph B Lillegard DOI: 10.19080/OAJS.2019.10.555788 Ex Vivo Gene Therapy: A “Cultured” Surgical Approach to Curing Inherited Liver Disease Caitlin J VanLith1, Robert A Kaiser1,2, Clara T Nicolas1 and Joseph B Lillegard1,2,3* 1Department of Surgery, Mayo Clinic, Rochester, MN, USA 2Midwest Fetal Care Center, Children’s Hospital of Minnesota, Minneapolis, MN, USA 3Pediatric Surgical Associates, Minneapolis, MN, USA Received: February 22, 2019; Published: March 21, 2019 *Corresponding author: Joseph B Lillegard, Midwest Fetal Care Center, Children’s Hospital of Minnesota, Minneapolis, Minnesota, USA and Mayo Clinic, Rochester, Minnesota, USA Introduction Inborn errors of metabolism (IEMs) are a group of inherited diseases caused by mutations in a single gene [1], many of which transplant remains the only curative option. Between 1988 and 2018, 12.8% of 17,009 pediatric liver transplants in the United States(see were primarily due to an inherited liver). disease. are identified in Table 1. Though individually rare, combined incidence is about 1 in 1,000 live births [2]. While maintenance www.optn.transplant.hrsa.gov/data/ Table 1: List of 35 of the most common Inborn Errors of Metabolism. therapies exist for some of these liver-related diseases, Inborn Error of Metabolism Abbreviation Hereditary Tyrosinemia type 1 HT1 Wilson Disease Wilson Glycogen Storage Disease 1 GSD1 Carnitine Palmitoyl Transferase Deficiency Type 2 CPT2 Glycogen Storage -

Module 01: Classification of Porphyria
MODULE 01 Classification of Porphyria Do not reprint, reproduce, modify or distribute this material without the prior written permission of Alnylam Pharmaceuticals. © 20120199 Alnylam PharmaceuticalsPharmaceuticals,, Inc.Inc. All rights reserved. -USA-00001-092018 1 Porphyria—A Rare Disease of Clinical Consequence • Porphyria is a group of at least 8 metabolic disorders1,2 – Each subtype of porphyria involves a genetic defect in a heme biosynthesis pathway enzyme1,2 – The subtypes of porphyria are associated with distinct signs and symptoms in patient populations that can differ by gender and age1,3 • Prevalence of some subtypes of porphyria may be higher than generally assumed3 Estimated Prevalence of Most Common Subtypes of Porphyria1,4 Estimated Prevalence Based on European Subtype of Porphyria and US Data Porphyria cutanea tarda (PCT) 1/10,000 (EU)1 0.118-1/20,000 (EU)1,4 Acute intermittent porphyria (AIP) 5/100,000 (US)1 Erythropoietic protoporphyria (EPP) 1/50,000-75,000 (EU)1 1. Ramanujam V-MS, Anderson KE. Curr Protoc Hum Genet. 2015;86:17.20.1-17.20.26. 2. Puy H et al. Lancet. 2010;375:924-937. 3. Bissell DM et al. N Engl J Med. 2017;377:862-872. 4. Elder G et al. J Inherit Metab Dis. 2013;36:848-857. Do not reprint, reproduce, modify or distribute this material without the prior written permission of Alnylam Pharmaceuticals. © 2019 Alnylam Pharmaceuticals, Inc. All rights reserved. -USA-00001-092018 2 Classification of Porphyria Porphyria can be classified in 2 major ways1,2: 1 According to major physiological sites: liver or 2 According to major clinical manifestations1,2 bone marrow1,2 • Heme precursors originate in either the liver or Acute Versus Photocutaneous Porphyria bone marrow, which are the tissues most • Major clinical manifestations are either neurovisceral active in heme biosynthesis1,2 symptoms (eg, severe, diffuse abdominal pain) associated with acute exacerbations or cutaneous lesions resulting from phototoxicity1,2 • Acute hepatic porphyria may be somewhat of a misnomer since the clinical features may be prolonged and chronic3 1. -

Variegate Porphyria with Coexistent Decrease in Porphobilinogen Deaminase Activity
Acta Derm Venereol 2001; 81: 356–359 CLINICAL REPORT Variegate Porphyria with Coexistent Decrease in Porphobilinogen Deaminase Activity GEORG WEINLICH1, MANFRED O. DOSS2, NORBERT SEPP1 and PETER FRITSCH1 1Department of Dermatology, University of Innsbruck, Innsbruck, Austria and 2Department of Clinical Biochemistry, University of Marburg, Marburg, Germany Variegate porphyria is a rare disease caused by a de ciency of deaminase. Its activity is reduced by about 50%, resulting in protoporphyrinogen oxidase. In most cases, the clinical ndings are varying degrees of overproduction and increased urinary excre- a combination of systemic symptoms similar to those occurring in tion of delta-aminolaevulinic acid (ALA) and PBG. In AIP, acute intermittent porphyria and cutaneous lesions indistinguishable skin changes are absent, but patients suVer from episodic from those of porphyria cutanea tarda. We report on a 24-year- central or peripheral nervous system and/or psychiatric symp- old woman with variegate porphyria who, after intake of lynestrenol, toms, or acute attacks of abdominal pain (2, 4). developed typical cutaneous lesions but no viscero-neurological Variegate porphyria (VP), a rare autosomal-dominant hep- symptoms. The diagnosis was based on the characteristic urinary atic porphyria due to a de ciency of protoporphyrinogen coproporphyrin and faecal protoporphyrin excretion patterns, and (PROTO) oxidase, the related gene (PPOX ) for which has the speci c peak of plasma uorescence at 626 nm in spectro uor- been located to chromosome 1q22-23 (5, 6), is characterized ometry. Biochemical analysis revealed that most of the family by both acute neurological symptoms as in AIP and skin members, though free of clinical symptoms, excrete porphyrin lesions indistinguishable from PCT; it is thus also termed metabolites in urine and stool similar to variegate porphyria, mixed porphyria (2, 7, 8). -

Advances in Hematology
ADVANCES IN HEMATOLOGY Current Developments in the Management of Hematologic Disorders Hematology Section Editor: Craig M. Kessler, MD What Hematologists Need to Know About Acute Hepatic Porphyria Manisha Balwani, MD, MS Associate Professor Department of Genetics and Genomic Sciences Icahn School of Medicine at Mount Sinai New York, New York H&O What is porphyria? H&O How common is acute hepatic porphyria? MB The porphyrias encompass a group of inherited MB Based on estimates from Western Europe, the metabolic disorders that result from a deficiency of one of combined prevalence of the acute hepatic porphyrias the enzymes in the heme biosynthetic pathway. These are among the white population is approximately 1 in genetic disorders; they can be inherited in an autosomal 200,000. These disorders are more common in certain dominant or recessive X-linked pattern; or they may be parts of the world because of founder mutations. For sporadic, as with porphyria cutanea tarda. example, the carrier frequency of acute intermittent porphyria is much higher in the Scandinavian countries, H&O How many types of porphyria exist? and variegate porphyria is much more common in South Africa.1 MB There are 8 different kinds of porphyria. These may be Even where the incidence is high, symptoms related classified as hepatic or erythropoietic based on the primary to these disorders remain rare. In fact, most patients who site of accumulation of porphyrins, but more commonly inherit a genetic change do not manifest symptoms of the they are classified clinically as acute or cutaneous. The acute disorder—the disease remains latent in a vast majority of hepatic porphyrias include acute intermittent porphyria these patients. -

Based Photodynamic Diagnosis of Malignant Brain Tumor: Molecular Design of ABCG2 Inhibitors
Pharmaceutics 2011 , 3, 615-635; doi:10.3390/pharmaceutics3030615 OPEN ACCESS pharmaceutics ISSN 1999-4923 www.mdpi.com/journal/pharmaceutics Review Transporter-Mediated Drug Interaction Strategy for 5-Aminolevulinic Acid (ALA)-Based Photodynamic Diagnosis of Malignant Brain Tumor: Molecular Design of ABCG2 Inhibitors Toshihisa Ishikawa 1,*, Kenkichi Takahashi 2, Naokado Ikeda 2, Yoshinaga Kajimoto 2, Yuichiro Hagiya 3, Shun-ichiro Ogura 3, Shin-ichi Miyatake 2 and Toshihiko Kuroiwa 2 1 Omics Science Center, RIKEN Yokohama Institute, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, 230-0045, Japan 2 Department of Neurosurgery, Osaka Medical College, Osaka 569-8686, Japan 3 Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, Yokohama 226-8501, Japan * Author to whom correspondence should be addressed: E-Mail: [email protected]; Tel.: +81-45-503-9222; Fax: +81-45-503-9216. Received: 19 July 2011; in revised form: 16 August 2011 / Accepted: 9 September 2011 / Published: 14 September 2011 Abstract: Photodynamic diagnosis (PDD) is a practical tool currently used in surgical operation of aggressive brain tumors, such as glioblastoma. PDD is achieved by a photon-induced physicochemical reaction which is induced by excitation of protoporphyrin IX (PpIX) exposed to light. Fluorescence-guided gross-total resection has recently been developed in PDD, where 5-aminolevulinic acid (ALA) or its ester is administered as the precursor of PpIX. ALA induces the accumulation of PpIX, a natural photo-sensitizer, in cancer cells. Recent studies provide evidence that adenosine triphosphate (ATP)-binding cassette (ABC) transporter ABCG2 plays a pivotal role in regulating the cellular accumulation of porphyrins in cancer cells and thereby affects the efficacy of PDD. -

Bilirubin Secretion, Jaundice and Evaluation of Liver Function
Bilirubin Secretion, Jaundice and Evaluation of Liver Function Howard J. Worman, M. D. Evaluation of Liver Disease and Hepatic Function History Physical Examination Laboratory Tests Sometimes Radiological/Nuclear Medicine Sometimes Liver Biopsy 1 Jaundice occurs as a result of excess bilirubin in the blood. It is a hallmark of liver disease but not always present in liver disease. Jaundice occurs when the liver fails to adequately secrete bilirubin from the blood into the bile. To understand how jaundice occurs, you must first understand bilirubin synthesis, metabolism and secretion. 2 Heme Oxygenase Schuller et al. Nature Structural Biology 6, 860 - 867 (1999) Bilivirdin Reductase Kikuchi et al. Nature Structural Biology 8, 221 - 225 (2001) 3 Bilirubin is frequently depicted as a linear tetrapyrrole. However, intramolecular hydrogen bonding fixes it in a rigid structure that blocks exposure of its polar groups to aqueous solvents, making it very insoluble in blood. 4 Bilirubin in Blood is Bound to Albumin: Uptake into Hepatocyte at Basolateral (Sinusoidal) Membrane Some OATP-C? bilirubin stored in cytosol bound to proteins Bilurubin UDP-glucuronosyltransferase is localized to the endo- plasmic reticulum; it catalyzes conjugation to a diglucuronide, making it more water soluble. A: Labeling of periphery of cell hepatocyte nucleus B: Labeling of ER with antibody to UDP-glucuronosyltransferase 5 Alternative RNA splicing of different first exons of UGT1 gives different isoforms with different substrate specificities, some for bilirubin and others -

Demystification of Chester Porphyria: a Nonsense Mutation in the Porphobilinogen Deaminase Gene
Physiol. Res. 55 (Suppl. 2): S137-S144, 2006 Demystification of Chester Porphyria: A Nonsense Mutation in the Porphobilinogen Deaminase Gene *P. POBLETE-GUTIÉRREZ1, *T. WIEDERHOLT2, 3, A. MARTINEZ-MIR4, H. F. MERK2, J. M. CONNOR5, A. M. CHRISTIANO4, 6, J. FRANK1,2,3 1Department of Dermatology, University Hospital Maastricht, The Netherlands, 2Department of Dermatology and Allergology and 3Porphyria Center, University Hospital of the RWTH Aachen, Germany, 4Department of Dermatology, Columbia University, New York, NY, USA, 5Institute of Medical Genetics, Yorkhill Hospitals, Glasgow, Scotland, 6Department of Genetics and Develop- ment, Columbia University, New York, NY, USA Received October 4, 2005 Accepted March 24, 2006 Summary The porphyrias arise from predominantly inherited catalytic deficiencies of specific enzymes in heme biosynthesis. All genes encoding these enzymes have been cloned and several mutations underlying the different types of porphyrias have been reported. Traditionally, the diagnosis of porphyria is made on the basis of clinical symptoms, characteristic biochemical findings, and specific enzyme assays. In some cases however, these diagnostic tools reveal overlapping findings, indicating the existence of dual porphyrias with two enzymes of heme biosynthesis being deficient simultane- ously. Recently, it was reported that the so-called Chester porphyria shows features of both variegate porphyria and acute intermittent porphyria. Linkage analysis revealed a novel chromosomal locus on chromosome 11 for the underly- ing genetic defect in this disease, suggesting that a gene that does not encode one of the enzymes of heme biosynthesis might be involved in the pathogenesis of the porphyrias. After excluding candidate genes within the linkage interval, we identified a nonsense mutation in the porphobilinogen deaminase gene on chromosome 11q23.3, which harbors the mutations causing acute intermittent porphyria, as the underlying genetic defect in Chester porphyria. -

A High Urinary Urobilinogen / Serum Total Bilirubin Ratio Reported in Abdominal Pain Patients Can Indicate Acute Hepatic Porphyria
A High Urinary Urobilinogen / Serum Total Bilirubin Ratio Reported in Abdominal Pain Patients Can Indicate Acute Hepatic Porphyria Chengyuan Song Shandong University Qilu Hospital Shaowei Sang Shandong University Qilu Hospital Yuan Liu ( [email protected] ) Shandong University Qilu Hospital https://orcid.org/0000-0003-4991-552X Research Keywords: acute hepatic porphyria, urinary urobilinogen, serum total bilirubin Posted Date: June 14th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-587707/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/10 Abstract Background: Due to its variable symptoms and nonspecic laboratory test results during routine examinations, acute hepatic porphyria (AHP) has always been a diagnostic dilemma for physicians. Misdiagnoses, missed diagnoses, and inappropriate treatments are very common. Correct diagnosis mainly depends on the detection of a high urinary porphobilinogen (PBG) level, which is not a routine test performed in the clinic and highly relies on the physician’s awareness of AHP. Therefore, identifying a more convenient indicator for use during routine examinations is required to improve the diagnosis of AHP. Results: In the present study, we retrospectively analyzed laboratory examinations in 12 AHP patients and 100 patients with abdominal pain of other causes as the control groups between 2015 and 2021. Compared with the control groups, AHP patients showed a signicantly higher urinary urobilinogen level during the urinalysis (P < 0.05). However, we showed that the higher urobilinogen level was caused by a false- positive result due to a higher level of urine PBG in the AHP patients. Hence, we used serum total bilirubin, an upstream substance of urinary urobilinogen synthesis, for calibration.