HHS Public Access Author Manuscript
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chromosomal Aberrations in Head and Neck Squamous Cell Carcinomas in Norwegian and Sudanese Populations by Array Comparative Genomic Hybridization
825-843 12/9/08 15:31 Page 825 ONCOLOGY REPORTS 20: 825-843, 2008 825 Chromosomal aberrations in head and neck squamous cell carcinomas in Norwegian and Sudanese populations by array comparative genomic hybridization ERIC ROMAN1,2, LEONARDO A. MEZA-ZEPEDA3, STINE H. KRESSE3, OLA MYKLEBOST3,4, ENDRE N. VASSTRAND2 and SALAH O. IBRAHIM1,2 1Department of Biomedicine, Faculty of Medicine and Dentistry, University of Bergen, Jonas Lies vei 91; 2Department of Oral Sciences - Periodontology, Faculty of Medicine and Dentistry, University of Bergen, Årstadveien 17, 5009 Bergen; 3Department of Tumor Biology, Institute for Cancer Research, Rikshospitalet-Radiumhospitalet Medical Center, Montebello, 0310 Oslo; 4Department of Molecular Biosciences, University of Oslo, Blindernveien 31, 0371 Oslo, Norway Received January 30, 2008; Accepted April 29, 2008 DOI: 10.3892/or_00000080 Abstract. We used microarray-based comparative genomic logical parameters showed little correlation, suggesting an hybridization to explore genome-wide profiles of chromosomal occurrence of gains/losses regardless of ethnic differences and aberrations in 26 samples of head and neck cancers compared clinicopathological status between the patients from the two to their pair-wise normal controls. The samples were obtained countries. Our findings indicate the existence of common from Sudanese (n=11) and Norwegian (n=15) patients. The gene-specific amplifications/deletions in these tumors, findings were correlated with clinicopathological variables. regardless of the source of the samples or attributed We identified the amplification of 41 common chromosomal carcinogenic risk factors. regions (harboring 149 candidate genes) and the deletion of 22 (28 candidate genes). Predominant chromosomal alterations Introduction that were observed included high-level amplification at 1q21 (harboring the S100A gene family) and 11q22 (including Head and neck squamous cell carcinoma (HNSCC), including several MMP family members). -
Download Validation Data
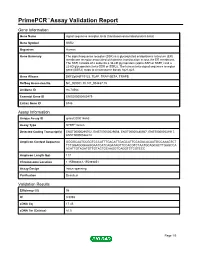
PrimePCR™Assay Validation Report Gene Information Gene Name signal sequence receptor, beta (translocon-associated protein beta) Gene Symbol SSR2 Organism Human Gene Summary The signal sequence receptor (SSR) is a glycosylated endoplasmic reticulum (ER) membrane receptor associated with protein translocation across the ER membrane. The SSR consists of 2 subunits a 34-kD glycoprotein (alpha-SSR or SSR1) and a 22-kD glycoprotein (beta-SSR or SSR2). The human beta-signal sequence receptor gene (SSR2) maps to chromosome bands 1q21-q23. Gene Aliases DKFZp686F19123, TLAP, TRAP-BETA, TRAPB RefSeq Accession No. NC_000001.10, NT_004487.19 UniGene ID Hs.74564 Ensembl Gene ID ENSG00000163479 Entrez Gene ID 6746 Assay Information Unique Assay ID qHsaCID0014663 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000295702, ENST00000529008, ENST00000480567, ENST00000531917, ENST00000526212 Amplicon Context Sequence GGGGCAATCCGGTCCCATTTGACATTGAGCATTCCAGACACAATGCCAAAGTCT TCTGGAGGGAAGGAATCATCAGATAGTTCCACGTCTAATGCAGCACTTGAGCCA ACATTGTAGATGTTGTACTGCAAGGTCAGGTCTCGTCCC Amplicon Length (bp) 117 Chromosome Location 1:155988061-155989851 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 98 R2 0.9998 cDNA Cq 17.45 cDNA Tm (Celsius) 81.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report SSR2, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Human Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AR50810PU-N SSR2 / TRAPB (18-149, His-tag) Human Protein Product data: Product Type: Recombinant Proteins Description: SSR2 / TRAPB (18-149, His-tag) human recombinant protein, 0.5 mg Species: Human Expression Host: E. coli Tag: His-tag Predicted MW: 16.8 kDa Concentration: lot specific Purity: >90% by SDS - PAGE Buffer: Presentation State: Purified State: Liquid purified protein Buffer System: 20 mM Tris-HCl buffer (pH8.0) containing 10% glycerol 0.1M NaCl Preparation: Liquid purified protein Protein Description: Recombinant human SSR2 protein, fused to His-tag at N-terminus, was expressed in E.coli and purified by using conventional chromatography techniques. Storage: Store undiluted at 2-8°C for one week or (in aliquots) at -20°C to -80°C for longer. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. RefSeq: NP_003136 Locus ID: 6746 UniProt ID: P43308 Cytogenetics: 1q22 Synonyms: HSD25; TLAP; TRAP-BETA; TRAPB Summary: The signal sequence receptor (SSR) is a glycosylated endoplasmic reticulum (ER) membrane receptor associated with protein translocation across the ER membrane. The SSR consists of 2 subunits, a 34-kD glycoprotein (alpha-SSR or SSR1) and a 22-kD glycoprotein (beta-SSR or SSR2). The human beta-signal sequence receptor gene (SSR2) maps to chromosome bands 1q21-q23. [provided by RefSeq, Jul 2008] This product is to be used for laboratory only. Not for diagnostic or therapeutic use. -

A Comparative Analysis of the Transcriptome Profiles of Liver and Muscle Tissue in Pigs Divergent for Feed Efficiency Stafford Vigors1, John V
Vigors et al. BMC Genomics (2019) 20:461 https://doi.org/10.1186/s12864-019-5740-z RESEARCH ARTICLE Open Access A comparative analysis of the transcriptome profiles of liver and muscle tissue in pigs divergent for feed efficiency Stafford Vigors1, John V. O’Doherty2, Kenneth Bryan2 and Torres Sweeney1* Abstract Background: The improvement of feed efficiency is a key economic goal within the pig production industry. The objective of this study was to examine transcriptomic differences in both the liver and muscle of pigs divergent for feed efficiency, thus improving our understanding of the molecular mechanisms influencing feed efficiency and enabling the identification of candidate biomarkers. Residual feed intake (RFI) was calculated for two populations of pigs from two different farms of origin/genotype. The 6 most efficient (LRFI) and 6 least efficient (HRFI) animals from each population were selected for further analysis of Longissimus Dorsi muscle (n = 22) and liver (n = 23). Transcriptomic data were generated from liver and muscle collected post-slaughter. Results: The transcriptomic data segregated based on the RFI value of the pig rather than genotype/farm of origin. A total of 6463 genes were identified as being differentially expressed (DE) in muscle, while 964 genes were identified as being DE in liver. Genes that were commonly DE between muscle and liver (n = 526) were used for the multi-tissue analysis. These 526 genes were associated with protein targeting to membrane, extracellular matrix organisation and immune function. In the muscle-only analysis, genes associated with RNA processing, protein synthesis and energy metabolism were down regulated in the LRFI animals while in the liver-only analysis, genes associated with cell signalling and lipid homeostasis were up regulated in the LRFI animals. -

Reverse Engineering of Modified Genes by Bayesian Network Analysis Defines Olecm Ular Determinants Critical to the Development of Glioblastoma Brian W
Florida International University FIU Digital Commons Robert Stempel College of Public Health & Social Environmental Health Sciences Work 5-30-2013 Reverse Engineering of Modified Genes by Bayesian Network Analysis Defines olecM ular Determinants Critical to the Development of Glioblastoma Brian W. Kunkle Department of Environmental Health Sciences, Florida International University Changwon Yoo Department of Biostatistics, Florida International University, [email protected] Deodutta Roy Department of Environmental Health Sciences, Florida International University, [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/eoh_fac Part of the Medicine and Health Sciences Commons Recommended Citation Kunkle BW, Yoo C, Roy D (2013) Reverse Engineering of Modified Genes by Bayesian Network Analysis Defines Molecular Determinants Critical to the Development of Glioblastoma. PLoS ONE 8(5): e64140. https://doi.org/10.1371/ journal.pone.0064140 This work is brought to you for free and open access by the Robert Stempel College of Public Health & Social Work at FIU Digital Commons. It has been accepted for inclusion in Environmental Health Sciences by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. Reverse Engineering of Modified Genes by Bayesian Network Analysis Defines Molecular Determinants Critical to the Development of Glioblastoma Brian W. Kunkle1, Changwon Yoo2, Deodutta Roy1* 1 Department of Environmental and Occupational Health, Florida International University, Miami, Florida, United States of America, 2 Department of Biostatistics, Florida International University, Miami, Florida, United States of America Abstract In this study we have identified key genes that are critical in development of astrocytic tumors. Meta-analysis of microarray studies which compared normal tissue to astrocytoma revealed a set of 646 differentially expressed genes in the majority of astrocytoma. -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis. -

Single-Cell Sequencing Reveals Clonally Expanded Plasma Cells During Chronic Viral Infection Produce Virus-Specific and Cross-Reactive Antibodies
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.29.428852; this version posted January 31, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Single-cell sequencing reveals clonally expanded plasma cells during chronic viral infection produce virus-specific and cross-reactive antibodies Daniel Neumeier1, Alessandro Pedrioli2 , Alessandro Genovese2, Ioana Sandu2, Roy Ehling1, Kai-Lin Hong1, Chrysa Papadopoulou1, Andreas Agrafiotis1, Raphael Kuhn1, Damiano Robbiani1, Jiami Han1, Laura Hauri1, Lucia Csepregi1, Victor Greiff3, Doron Merkler4,5, Sai T. Reddy1,*, Annette Oxenius2,*, Alexander Yermanos1,2,4,* 1Department of Biosystems Science and Engineering, ETH Zurich, Basel, Switzerland 2Institute of Microbiology, ETH Zurich, Zurich, Switzerland 3Department of Immunology, University of Oslo, Oslo, Norway 4Department of Pathology and Immunology, University of Geneva, Geneva, Switzerland 5Division of Clinical Pathology, Geneva University Hospital, Geneva, Switzerland *Correspondence: [email protected] ; [email protected] ; [email protected] Graphical abstract. Single-cell sequencing reveals clonally expanded plasma cells during chronic viral infection produce virus-specific and cross-reactive antibodies. bioRxiv preprint doi: https://doi.org/10.1101/2021.01.29.428852; this version posted January 31, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Neumeier et al., Abstract Plasma cells and their secreted antibodies play a central role in the long-term protection against chronic viral infection. -

Peripheral Nerve Single-Cell Analysis Identifies Mesenchymal Ligands That Promote Axonal Growth
Research Article: New Research Development Peripheral Nerve Single-Cell Analysis Identifies Mesenchymal Ligands that Promote Axonal Growth Jeremy S. Toma,1 Konstantina Karamboulas,1,ª Matthew J. Carr,1,2,ª Adelaida Kolaj,1,3 Scott A. Yuzwa,1 Neemat Mahmud,1,3 Mekayla A. Storer,1 David R. Kaplan,1,2,4 and Freda D. Miller1,2,3,4 https://doi.org/10.1523/ENEURO.0066-20.2020 1Program in Neurosciences and Mental Health, Hospital for Sick Children, 555 University Avenue, Toronto, Ontario M5G 1X8, Canada, 2Institute of Medical Sciences University of Toronto, Toronto, Ontario M5G 1A8, Canada, 3Department of Physiology, University of Toronto, Toronto, Ontario M5G 1A8, Canada, and 4Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5G 1A8, Canada Abstract Peripheral nerves provide a supportive growth environment for developing and regenerating axons and are es- sential for maintenance and repair of many non-neural tissues. This capacity has largely been ascribed to paracrine factors secreted by nerve-resident Schwann cells. Here, we used single-cell transcriptional profiling to identify ligands made by different injured rodent nerve cell types and have combined this with cell-surface mass spectrometry to computationally model potential paracrine interactions with peripheral neurons. These analyses show that peripheral nerves make many ligands predicted to act on peripheral and CNS neurons, in- cluding known and previously uncharacterized ligands. While Schwann cells are an important ligand source within injured nerves, more than half of the predicted ligands are made by nerve-resident mesenchymal cells, including the endoneurial cells most closely associated with peripheral axons. At least three of these mesen- chymal ligands, ANGPT1, CCL11, and VEGFC, promote growth when locally applied on sympathetic axons. -

Exploring the Role of Endoplasmic Reticulum Stress in Hepatocellular Carcinoma Through Mining of the Human Protein Atlas
biology Article Exploring the Role of Endoplasmic Reticulum Stress in Hepatocellular Carcinoma through mining of the Human Protein Atlas Nataša Pavlovi´cand Femke Heindryckx * Department of Medical Cell Biology, Uppsala University, 751 23 Uppsala, Sweden; [email protected] * Correspondence: [email protected] Simple Summary: Hepatocellular carcinoma is a highly deadly primary liver cancer. It is usually diagnosed at a late stage, when therapeutic options are scarce, and the lack of predictive biomarkers poses a challenge for early detection. A known hallmark of hepatocellular carcinoma is the accu- mulation of misfolded proteins in the endoplasmic reticulum (ER), known as ER-stress. Growing experimental evidence suggests that ER-stress is involved in liver cancer initiation and progression. However, it remains unclear if ER-stress markers can be used as therapeutic targets or biomarkers for patients with liver cancer. In this study, we evaluated the prognostic value of proteins involved in managing ER-stress in liver cancer by mining a publicly available patient-derived database, the Human Protein Atlas. We thereby identified 44 ER-stress-associated proteins as prognostic markers in liver cancer. Furthermore, we discussed the expression of these markers in relation to disease stage, age, sex, ethnicity, and tissue localization. Abstract: Endoplasmic reticulum (ER) stress and actors of unfolded protein response (UPR) have Citation: Pavlovi´c,N.; Heindryckx, F. emerged as key hallmarks of hepatocarcinogenesis. Numerous reports have shown that the main Exploring the Role of Endoplasmic actors in the UPR pathways are upregulated in HCC and contribute to the different facets of tumor Reticulum Stress in Hepatocellular initiation and disease progression. -

Mutation of Mouse Samd4 Causes Leanness, Myopathy, Uncoupled Mitochondrial Respiration, and Dysregulated Mtorc1 Signaling
Mutation of mouse Samd4 causes leanness, myopathy, uncoupled mitochondrial respiration, and dysregulated mTORC1 signaling Zhe Chena, William Hollandb, John M. Sheltonc, Aktar Alid, Xiaoming Zhana, Sungyong Wona,1, Wataru Tomisatoa,2, Chen Liue, Xiaohong Lia, Eva Marie Y. Morescoa, and Bruce Beutlera,3 aCenter for Genetics of Host Defense, bDepartment of Internal Medicine, Touchstone Diabetes Center, cDepartment of Internal Medicine, Division of Cardiology, dDepartment of Internal Medicine, and eDepartment of Internal Medicine, Division of Hypothalamic Research, University of Texas Southwestern Medical Center, Dallas, TX 75390 Contributed by Bruce Beutler, April 14, 2014 (sent for review March 12, 2014) Sterile alpha motif domain containing protein 4 (Samd4) is an RNA span compared with control littermates, with deaths beginning at binding protein that mediates translational repression. We iden- approximately 3 mo of age (Fig. 1B). tified a Samd4 missense mutation, designated supermodel, that Male and female homozygous spmd mice fed standard rodent caused leanness and kyphosis associated with myopathy and adi- chow exhibited lower body weight, body mass index, and body pocyte defects in C57BL/6J mice. The supermodel mutation pro- length relative to wild-type littermates (Fig. 1C and Table S1). tected homozygous mice from high fat diet-induced obesity, likely When challenged with a high fat diet (HFD), wild-type mice in- < by promoting enhanced energy expenditure through uncoupled creased their initial body weight 1.5-fold (males; P 0.01) or < mitochondrial respiration. Glucose tolerance was impaired due to 1.8-fold (females; P 0.01) over a 6-mo period, whereas spmd homozygotes increased it by only 1.3-fold (males; P = 0.028) or diminished insulin release in homozygous mutant mice. -

Murine Perinatal Beta Cell Proliferation and the Differentiation of Human Stem Cell Derived Insulin Expressing Cells Require NEUROD1
Page 1 of 105 Diabetes Murine perinatal beta cell proliferation and the differentiation of human stem cell derived insulin expressing cells require NEUROD1 Anthony I. Romer,1,2 Ruth A. Singer1,3, Lina Sui2, Dieter Egli,2* and Lori Sussel1,4* 1Department of Genetics and Development, Columbia University, New York, NY 10032, USA 2Department of Pediatrics, Columbia University, New York, NY 10032, USA 3Integrated Program in Cellular, Molecular and Biomedical Studies, Columbia University, New York, NY 10032, USA 4Department of Pediatrics, University of Colorado Denver School of Medicine, Denver, CO 80045, USA *Co-Corresponding Authors Dieter Egli 1150 St. Nicholas Avenue New York, NY 10032 [email protected] Lori Sussel 1775 Aurora Ct. Aurora, CO 80045 [email protected] Word Count: Abstract= 149; Body= 4773 Total Paper Figures= 7, Total Supplemental Tables= 4, Total Supplemental Figures= 5 Diabetes Publish Ahead of Print, published online September 13, 2019 Diabetes Page 2 of 105 Abstract Inactivation of the β cell transcription factor NEUROD1 causes diabetes in mice and humans. In this study, we uncovered novel functions of Neurod1 during murine islet cell development and during the differentiation of human embryonic stem cells (HESCs) into insulin-producing cells. In mice, we determined that Neurod1 is required for perinatal proliferation of alpha and beta cells. Surprisingly, apoptosis only makes a minor contribution to beta cell loss when Neurod1 is deleted. Inactivation of NEUROD1 in HESCs severely impaired their differentiation from pancreatic progenitors into insulin expressing (HESC-beta) cells; however survival or proliferation was not affected at the time points analyzed. NEUROD1 was also required in HESC-beta cells for the full activation of an essential beta cell transcription factor network.