Increased DNA Methylation Variability in Type 1 Diabetes Across Three Immune Effector Cell Types
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lower Genomic Stability of Induced Pluripotent Stem Cells Reflects
Zhang et al. Cancer Commun (2018) 38:49 https://doi.org/10.1186/s40880-018-0313-0 Cancer Communications ORIGINAL ARTICLE Open Access Lower genomic stability of induced pluripotent stem cells refects increased non‑homologous end joining Minjie Zhang1,2†, Liu Wang3†, Ke An1,2†, Jun Cai1, Guochao Li1,2, Caiyun Yang1, Huixian Liu1, Fengxia Du1, Xiao Han1,2, Zilong Zhang1,2, Zitong Zhao1,2, Duanqing Pei4, Yuan Long5, Xin Xie5, Qi Zhou3 and Yingli Sun1* Abstract Background: Induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs) share many common features, including similar morphology, gene expression and in vitro diferentiation profles. However, genomic stability is much lower in iPSCs than in ESCs. In the current study, we examined whether changes in DNA damage repair in iPSCs are responsible for their greater tendency towards mutagenesis. Methods: Mouse iPSCs, ESCs and embryonic fbroblasts were exposed to ionizing radiation (4 Gy) to introduce dou- ble-strand DNA breaks. At 4 h later, fdelity of DNA damage repair was assessed using whole-genome re-sequencing. We also analyzed genomic stability in mice derived from iPSCs versus ESCs. Results: In comparison to ESCs and embryonic fbroblasts, iPSCs had lower DNA damage repair capacity, more somatic mutations and short indels after irradiation. iPSCs showed greater non-homologous end joining DNA repair and less homologous recombination DNA repair. Mice derived from iPSCs had lower DNA damage repair capacity than ESC-derived mice as well as C57 control mice. Conclusions: The relatively low genomic stability of iPSCs and their high rate of tumorigenesis in vivo appear to be due, at least in part, to low fdelity of DNA damage repair. -

In Silico Prediction of High-Resolution Hi-C Interaction Matrices
ARTICLE https://doi.org/10.1038/s41467-019-13423-8 OPEN In silico prediction of high-resolution Hi-C interaction matrices Shilu Zhang1, Deborah Chasman 1, Sara Knaack1 & Sushmita Roy1,2* The three-dimensional (3D) organization of the genome plays an important role in gene regulation bringing distal sequence elements in 3D proximity to genes hundreds of kilobases away. Hi-C is a powerful genome-wide technique to study 3D genome organization. Owing to 1234567890():,; experimental costs, high resolution Hi-C datasets are limited to a few cell lines. Computa- tional prediction of Hi-C counts can offer a scalable and inexpensive approach to examine 3D genome organization across multiple cellular contexts. Here we present HiC-Reg, an approach to predict contact counts from one-dimensional regulatory signals. HiC-Reg pre- dictions identify topologically associating domains and significant interactions that are enri- ched for CCCTC-binding factor (CTCF) bidirectional motifs and interactions identified from complementary sources. CTCF and chromatin marks, especially repressive and elongation marks, are most important for HiC-Reg’s predictive performance. Taken together, HiC-Reg provides a powerful framework to generate high-resolution profiles of contact counts that can be used to study individual locus level interactions and higher-order organizational units of the genome. 1 Wisconsin Institute for Discovery, 330 North Orchard Street, Madison, WI 53715, USA. 2 Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Madison, WI 53715, USA. *email: [email protected] NATURE COMMUNICATIONS | (2019) 10:5449 | https://doi.org/10.1038/s41467-019-13423-8 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-019-13423-8 he three-dimensional (3D) organization of the genome has Results Temerged as an important component of the gene regulation HiC-Reg for predicting contact count using Random Forests. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

Chromosomal Aberrations in Head and Neck Squamous Cell Carcinomas in Norwegian and Sudanese Populations by Array Comparative Genomic Hybridization
825-843 12/9/08 15:31 Page 825 ONCOLOGY REPORTS 20: 825-843, 2008 825 Chromosomal aberrations in head and neck squamous cell carcinomas in Norwegian and Sudanese populations by array comparative genomic hybridization ERIC ROMAN1,2, LEONARDO A. MEZA-ZEPEDA3, STINE H. KRESSE3, OLA MYKLEBOST3,4, ENDRE N. VASSTRAND2 and SALAH O. IBRAHIM1,2 1Department of Biomedicine, Faculty of Medicine and Dentistry, University of Bergen, Jonas Lies vei 91; 2Department of Oral Sciences - Periodontology, Faculty of Medicine and Dentistry, University of Bergen, Årstadveien 17, 5009 Bergen; 3Department of Tumor Biology, Institute for Cancer Research, Rikshospitalet-Radiumhospitalet Medical Center, Montebello, 0310 Oslo; 4Department of Molecular Biosciences, University of Oslo, Blindernveien 31, 0371 Oslo, Norway Received January 30, 2008; Accepted April 29, 2008 DOI: 10.3892/or_00000080 Abstract. We used microarray-based comparative genomic logical parameters showed little correlation, suggesting an hybridization to explore genome-wide profiles of chromosomal occurrence of gains/losses regardless of ethnic differences and aberrations in 26 samples of head and neck cancers compared clinicopathological status between the patients from the two to their pair-wise normal controls. The samples were obtained countries. Our findings indicate the existence of common from Sudanese (n=11) and Norwegian (n=15) patients. The gene-specific amplifications/deletions in these tumors, findings were correlated with clinicopathological variables. regardless of the source of the samples or attributed We identified the amplification of 41 common chromosomal carcinogenic risk factors. regions (harboring 149 candidate genes) and the deletion of 22 (28 candidate genes). Predominant chromosomal alterations Introduction that were observed included high-level amplification at 1q21 (harboring the S100A gene family) and 11q22 (including Head and neck squamous cell carcinoma (HNSCC), including several MMP family members). -

CLASP2 Antibody Product Type
PRODUCT INFORMATION Product name: CLASP2 antibody Product type: Primary antibodies Description: Rabbit polyclonal to CLASP2 Immunogen:3 synthetic peptides (human) conjugated to KLH Reacts with:Hu, Ms Tested applications:ELISA, WB and IF GENE INFORMATION Gene Symbol: CLASP2 Gene Name:cytoplasmic linker associated protein 2 Ensembl ID:ENSG00000163539 Entrez GeneID:23122 GenBank Accession number:AB014527 Swiss-Prot:O75122 Molecular weight of CLASP2: 165.9 & 108.6kDa Function:Microtubule plus-end tracking protein that promotes the stabilization of dynamic microtubules. Involved in the nucleation of noncentrosomal microtubules originating from the trans-Golgi network (TGN). Required for the polarization of the cytoplasmic microtubule arrays in migrating cells towards the leading edge of the cell. May act at the cell cortex to enhance the frequency of rescue of depolymerizing microtubules by attaching their plus- ends to cortical platforms composed of ERC1 and PHLDB2. This cortical microtubule stabilizing activity is regulated at least in part by phosphatidylinositol 3-kinase signaling. Also performs a similar stabilizing function at the kinetochore which is essential for the bipolar alignment of chromosomes on the mitotic spindle. Acts as a mediator of ERBB2- dependent stabilization of microtubules at the cell cortex. Expected subcellular localization:Cytoplasm › cytoskeleton. Cytoplasm › cytoskeleton › microtubule organizing center › centrosome. Chromosome › centromere › kinetochore. Cytoplasm › cytoskeleton › spindle. Golgi apparatus. Golgi apparatus › trans-Golgi network. Cell membrane. Cell projection › ruffle membrane. Note: Localizes to microtubule plus ends. Localizes to centrosomes, kinetochores and the mitotic spindle from prometaphase. Subsequently localizes to the spindle midzone from anaphase and to the midbody from telophase. In migrating cells localizes to the plus ends of microtubules within the cell body and to the entire microtubule lattice within the lamella. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Misplaced Lncrna Causes Brachydactyly in Humans
A misplaced lncRNA causes brachydactyly in humans Philipp G. Maass, … , Friedrich C. Luft, Sylvia Bähring J Clin Invest. 2012;122(11):3990-4002. https://doi.org/10.1172/JCI65508. Research Article Translocations are chromosomal rearrangements that are frequently associated with a variety of disease states and developmental disorders. We identified 2 families with brachydactyly type E (BDE) resulting from different translocations affecting chromosome 12p. Both translocations caused downregulation of the parathyroid hormone-like hormone (PTHLH) gene by disrupting the cis-regulatory landscape. Using chromosome conformation capturing, we identified a regulator on chromosome 12q that interacts in cis with PTHLH over a 24.4-megabase distance and in trans with the sex- determining region Y-box 9 (SOX9) gene on chromosome 17q. The element also harbored a long noncoding RNA (lncRNA). Silencing of the lncRNA, PTHLH, or SOX9 revealed a feedback mechanism involving an expression-dependent network in humans. In the BDE patients, the human lncRNA was upregulated by the disrupted chromosomal association. Moreover, the lncRNA occupancy at the PTHLH locus was reduced. Our results document what we believe to be a novel in cis– and in trans–acting DNA and lncRNA regulatory feedback element that is reciprocally regulated by coding genes. Furthermore, our findings provide a systematic and combinatorial view of how enhancers encoding lncRNAs may affect gene expression in normal development. Find the latest version: https://jci.me/65508/pdf Research article Related Commentary, page 3837 A misplaced lncRNA causes brachydactyly in humans Philipp G. Maass,1,2 Andreas Rump,3 Herbert Schulz,2 Sigmar Stricker,4 Lisanne Schulze,1,2 Konrad Platzer,3 Atakan Aydin,1,2 Sigrid Tinschert,3 Mary B. -

Mouse Phldb2 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Phldb2 Knockout Project (CRISPR/Cas9) Objective: To create a Phldb2 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Phldb2 gene (NCBI Reference Sequence: NM_001252442 ; Ensembl: ENSMUSG00000033149 ) is located on Mouse chromosome 16. 19 exons are identified, with the ATG start codon in exon 2 and the TAG stop codon in exon 19 (Transcript: ENSMUST00000076333). Exon 2 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a conditional allele activated in neurons exhibit impaired LTP. Exon 2 starts from the coding region. Exon 2 covers 33.87% of the coding region. The size of effective KO region: ~1337 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 19 Legends Exon of mouse Phldb2 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 2 is aligned with itself to determine if there are tandem repeats. -
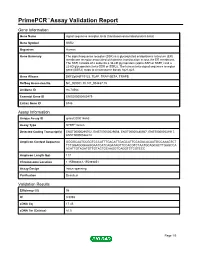
Download Validation Data
PrimePCR™Assay Validation Report Gene Information Gene Name signal sequence receptor, beta (translocon-associated protein beta) Gene Symbol SSR2 Organism Human Gene Summary The signal sequence receptor (SSR) is a glycosylated endoplasmic reticulum (ER) membrane receptor associated with protein translocation across the ER membrane. The SSR consists of 2 subunits a 34-kD glycoprotein (alpha-SSR or SSR1) and a 22-kD glycoprotein (beta-SSR or SSR2). The human beta-signal sequence receptor gene (SSR2) maps to chromosome bands 1q21-q23. Gene Aliases DKFZp686F19123, TLAP, TRAP-BETA, TRAPB RefSeq Accession No. NC_000001.10, NT_004487.19 UniGene ID Hs.74564 Ensembl Gene ID ENSG00000163479 Entrez Gene ID 6746 Assay Information Unique Assay ID qHsaCID0014663 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000295702, ENST00000529008, ENST00000480567, ENST00000531917, ENST00000526212 Amplicon Context Sequence GGGGCAATCCGGTCCCATTTGACATTGAGCATTCCAGACACAATGCCAAAGTCT TCTGGAGGGAAGGAATCATCAGATAGTTCCACGTCTAATGCAGCACTTGAGCCA ACATTGTAGATGTTGTACTGCAAGGTCAGGTCTCGTCCC Amplicon Length (bp) 117 Chromosome Location 1:155988061-155989851 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 98 R2 0.9998 cDNA Cq 17.45 cDNA Tm (Celsius) 81.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report SSR2, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Role of RUNX1 in Aberrant Retinal Angiogenesis Jonathan D
Page 1 of 25 Diabetes Identification of RUNX1 as a mediator of aberrant retinal angiogenesis Short Title: Role of RUNX1 in aberrant retinal angiogenesis Jonathan D. Lam,†1 Daniel J. Oh,†1 Lindsay L. Wong,1 Dhanesh Amarnani,1 Cindy Park- Windhol,1 Angie V. Sanchez,1 Jonathan Cardona-Velez,1,2 Declan McGuone,3 Anat O. Stemmer- Rachamimov,3 Dean Eliott,4 Diane R. Bielenberg,5 Tave van Zyl,4 Lishuang Shen,1 Xiaowu Gai,6 Patricia A. D’Amore*,1,7 Leo A. Kim*,1,4 Joseph F. Arboleda-Velasquez*1 Author affiliations: 1Schepens Eye Research Institute/Massachusetts Eye and Ear, Department of Ophthalmology, Harvard Medical School, 20 Staniford St., Boston, MA 02114 2Universidad Pontificia Bolivariana, Medellin, Colombia, #68- a, Cq. 1 #68305, Medellín, Antioquia, Colombia 3C.S. Kubik Laboratory for Neuropathology, Massachusetts General Hospital, 55 Fruit St., Boston, MA 02114 4Retina Service, Massachusetts Eye and Ear Infirmary, Department of Ophthalmology, Harvard Medical School, 243 Charles St., Boston, MA 02114 5Vascular Biology Program, Boston Children’s Hospital, Department of Surgery, Harvard Medical School, 300 Longwood Ave., Boston, MA 02115 6Center for Personalized Medicine, Children’s Hospital Los Angeles, Los Angeles, 4650 Sunset Blvd, Los Angeles, CA 90027, USA 7Department of Pathology, Harvard Medical School, 25 Shattuck St., Boston, MA 02115 Corresponding authors: Joseph F. Arboleda-Velasquez: [email protected] Ph: (617) 912-2517 Leo Kim: [email protected] Ph: (617) 912-2562 Patricia D’Amore: [email protected] Ph: (617) 912-2559 Fax: (617) 912-0128 20 Staniford St. Boston MA, 02114 † These authors contributed equally to this manuscript Word Count: 1905 Tables and Figures: 4 Diabetes Publish Ahead of Print, published online April 11, 2017 Diabetes Page 2 of 25 Abstract Proliferative diabetic retinopathy (PDR) is a common cause of blindness in the developed world’s working adult population, and affects those with type 1 and type 2 diabetes mellitus. -

Genome‑Wide Investigation of the Clinical Implications and Molecular Mechanism of Long Noncoding RNA LINC00668 and Protein‑Coding Genes in Hepatocellular Carcinoma
860 INTERNATIONAL JOURNAL OF ONCOLOGY 55: 860-878 2019 Genome‑wide investigation of the clinical implications and molecular mechanism of long noncoding RNA LINC00668 and protein‑coding genes in hepatocellular carcinoma XIANGKUN WANG1, XIN ZHOU1, JUNQI LIU1, ZHENGQIAN LIU1, LINBO ZHANG2, YIZHEN GONG3, JIANLU HUANG4, LONG YU1,5, QIAOQI WANG6, CHENGKUN YANG1, XIWEN LIAO1, TINGDONG YU1, CHUANGYE HAN1, GUANGZHI ZHU1, XINPING YE1 and TAO PENG1 Department of 1Hepatobiliary Surgery, 2Health Management and Division of Physical Examination and 3Colorectal and Anal Surgery, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region 530021; 4Department of Hepatobiliary Surgery, Third Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region 530031; 5Department of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450000; 6Department of Medical Cosmetology, The Second Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region 530000, P.R. China Received April 12, 2019; Accepted July 31, 2019 DOI: 10.3892/ijo.2019.4858 Abstract. Hepatocellular carcinoma (HCC) is one of the clinical factors and PCGs were used to construct a nomo- leading causes of tumor-related mortalities worldwide. gram for predicting prognosis in HCC. A Connectivity Map Long noncoding RNAs have been reported to be associ- was constructed to identify candidate target drugs for HCC. ated with tumor initiation, progression and prognosis. The The top 10 PCGs identified were: Pyrimidineregic receptor present study aimed to explore the association between long P2Y4 (P2RY4), signal peptidase complex subunit 2 (SPCS2), noncoding RNA LINC00668 and its co-expression correlated family with sequence similarity 86 member C1 (FAM86C1), protein-coding genes (PCGs) in HCC.