Domain Dynamics and Control of Electron Flux of NADPH Cytochrome P450 Oxidoreductase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amplitaq and Amplitaq Gold DNA Polymerase
AmpliTaq and AmpliTaq Gold DNA Polymerase The Most Referenced Brand of DNA Polymerase in the World Date: 2005-05 Notes: Authors are listed alphabetically J. Biol. Chem. (223) Ezoe, S., I. Matsumura, et al. (2005). "GATA Transcription Factors Inhibit Cytokine-dependent Growth and Survival of a Hematopoietic Cell Line through the Inhibition of STAT3 Activity." J. Biol. Chem. 280(13): 13163-13170. http://www.jbc.org/cgi/content/abstract/280/13/13163 Although GATA-1 and GATA-2 were shown to be essential for the development of hematopoietic cells by gene targeting experiments, they were also reported to inhibit the growth of hematopoietic cells. Therefore, in this study, we examined the effects of GATA-1 and GATA-2 on cytokine signals. A tamoxifen-inducible form of GATA-1 (GATA-1/ERT) showed a minor inhibitory effect on interleukin-3 (IL-3)-dependent growth of an IL-3-dependent cell line Ba/F3. On the other hand, it drastically inhibited TPO-dependent growth and gp130-mediated growth/survival of Ba/F3. Similarly, an estradiol-inducible form of GATA-2 (GATA-2/ER) disrupted thrombopoietin (TPO)-dependent growth and gp130-mediated growth/survival of Ba/F3. As for this mechanism, we found that both GATA-1 and GATA-2 directly bound to STAT3 both in vitro and in vivo and inhibited its DNA-binding activity in gel shift assays and chromatin immunoprecipitation assays, whereas they hardly affected STAT5 activity. In addition, endogenous GATA-1 was found to interact with STAT3 in normal megakaryocytes, suggesting that GATA-1 may inhibit STAT3 activity in normal hematopoietic cells. -

Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae
Journal of Fungi Article Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae Mohammad Sayari 1,2,*, Magrieta A. van der Nest 1,3, Emma T. Steenkamp 1, Saleh Rahimlou 4 , Almuth Hammerbacher 1 and Brenda D. Wingfield 1 1 Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (A.H.); brenda.wingfi[email protected] (B.D.W.) 2 Department of Plant Science, University of Manitoba, 222 Agriculture Building, Winnipeg, MB R3T 2N2, Canada 3 Biotechnology Platform, Agricultural Research Council (ARC), Onderstepoort Campus, Pretoria 0110, South Africa 4 Department of Mycology and Microbiology, University of Tartu, 14A Ravila, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Fax: +1-204-474-7528 Abstract: Terpenes represent the biggest group of natural compounds on earth. This large class of organic hydrocarbons is distributed among all cellular organisms, including fungi. The different classes of terpenes produced by fungi are mono, sesqui, di- and triterpenes, although triterpene ergosterol is the main sterol identified in cell membranes of these organisms. The availability of genomic data from members in the Ceratocystidaceae enabled the detection and characterization of the genes encoding the enzymes in the mevalonate and ergosterol biosynthetic pathways. Using Citation: Sayari, M.; van der Nest, a bioinformatics approach, fungal orthologs of sterol biosynthesis genes in nine different species M.A.; Steenkamp, E.T.; Rahimlou, S.; of the Ceratocystidaceae were identified. -

CYP2A6) by P53
Transcriptional Regulation of Human Stress Responsive Cytochrome P450 2A6 (CYP2A6) by p53 Hao Hu M.Biotech. (Biotechnology) 2012 The University of Queensland B.B.A. 2009 University of Electronic Science and Technology of China B.Sc. (Pharmacy) 2009 University of Electronic Science and Technology of China A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2016 School of Medicine ABSTRACT Human cytochrome P450 (CYP) 2A6 is highly expressed in the liver and the encoding gene is regulated by various stress activated transcription factors, such as the nuclear factor (erythroid-derived 2)-like 2 (Nrf-2). Unlike the other xenobiotic metabolising CYP enzymes (XMEs), CYP2A6 only plays a minor role in xenobiotic metabolism. The CYP2A6 is highly induced by multiple forms of cellular stress conditions, where XMEs expression is normally inhibited. Recent findings suggest that the CYP2A6 plays an important role in regulating BR homeostasis. A computer based sequence analysis on the 3 kb proximate CYP2A6 promoter revealed several putative binding sites for p53, a protein that mediates regulation of antioxidant and apoptosis pathways. In this study, the role of p53 in CYP2A6 gene regulation is demonstrated. The site closest to transcription start site (TSS) is highly homologous with the p53 consensus sequence. The p53 responsiveness of this site was confirmed by transfections with various stepwise deleted of CYP2A6-5’-Luc constructs containing the putative p53RE. Deletion of the putative p53RE resulted in a total abolishment of p53 responsiveness of CYP2A6 promoter. Specific binding of p53 to the putative p53RE was detected by electrophoresis mobility shift assay. -

Characterisation of Bilirubin Metabolic Pathway in Hepatic Mitochondria Siti Nur Fadzilah Muhsain M.Sc
Characterisation of Bilirubin Metabolic Pathway in Hepatic Mitochondria Siti Nur Fadzilah Muhsain M.Sc. (Medical Research) 2005 Universiti Sains Malaysia Postgrad. Dip. (Toxicology) 2003 University of Surrey B.Sc.(Biomed. Sc.) 2000 Universiti Putra Malaysia A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2014 School of Medicine ABSTRACT Bilirubin (BR), a toxic waste product of degraded haem, is a potent antioxidant at physiological concentrations. To achieve the maximum benefit of BR, its intracellular level needs to be carefully regulated. A system comprising of two enzymes, haem oxygenase-1 (HMOX1) and cytochrome P450 2A5 (CYP2A5) exists in the endoplasmic reticulum (ER), responsible for regulating BR homeostasis. This system is induced in response to oxidative stress. In this thesis, oxidative stress caused accumulation of these enzymes in mitochondria — major producers and targets of reactive oxygen species (ROS) — is demonstrated. To understand the significance of this intracellular targeting, properties of microsomal and mitochondrial BR metabolising enzymes were compared and the capacity of mitochondrial CYP2A5 to oxidise BR in response to oxidative stress is reported. Microsomes and mitochondrial fractions were isolated from liver homogenates of DBA/2J mice, administered with sub-toxic dose of pyrazole, an oxidant stressor. The purity of extracted organelles was determined by analysing the expressions and activities of their respective marker enzymes. HMOX1 and CYP2A5 were significantly increased in microsomes and even more so in mitochondria in response to pyrazole-induced oxidative stress. By contrast, the treatment did not increase either microsomes or mitochondrial Uridine-diphosphate-glucuronosyltransferase 1A1 (UGT1A1), the sole enzyme that catalyses BR elimination through glucuronidation. -

The Impact of Transcription on Metabolism in Prostate and Breast Cancers
25 9 Endocrine-Related N Poulose et al. From hormones to fats and 25:9 R435–R452 Cancer back REVIEW The impact of transcription on metabolism in prostate and breast cancers Ninu Poulose1, Ian G Mills1,2,* and Rebecca E Steele1,* 1Centre for Cancer Research and Cell Biology, Queen’s University of Belfast, Belfast, UK 2Nuffield Department of Surgical Sciences, John Radcliffe Hospital, University of Oxford, Oxford, UK Correspondence should be addressed to I G Mills: [email protected] *(I G Mills and R E Steele contributed equally to this work) Abstract Metabolic dysregulation is regarded as an important driver in cancer development and Key Words progression. The impact of transcriptional changes on metabolism has been intensively f androgen studied in hormone-dependent cancers, and in particular, in prostate and breast cancer. f androgen receptor These cancers have strong similarities in the function of important transcriptional f breast drivers, such as the oestrogen and androgen receptors, at the level of dietary risk and f oestrogen epidemiology, genetics and therapeutically. In this review, we will focus on the function f endocrine therapy of these nuclear hormone receptors and their downstream impact on metabolism, with a resistance particular focus on lipid metabolism. We go on to discuss how lipid metabolism remains dysregulated as the cancers progress. We conclude by discussing the opportunities that this presents for drug repurposing, imaging and the development and testing of new Endocrine-Related Cancer therapeutics and treatment combinations. (2018) 25, R435–R452 Introduction: prostate and breast cancer Sex hormones act through nuclear hormone receptors Early-stage PCa is dependent on androgens for survival and induce distinct transcriptional programmes essential and can be treated by androgen deprivation therapy; to male and female physiology. -

Heme Oxygenase-1 Regulates Mitochondrial Quality Control in the Heart
RESEARCH ARTICLE Heme oxygenase-1 regulates mitochondrial quality control in the heart Travis D. Hull,1,2 Ravindra Boddu,1,3 Lingling Guo,2,3 Cornelia C. Tisher,1,3 Amie M. Traylor,1,3 Bindiya Patel,1,4 Reny Joseph,1,3 Sumanth D. Prabhu,1,4,5 Hagir B. Suliman,6 Claude A. Piantadosi,7 Anupam Agarwal,1,3,5 and James F. George2,3,4 1Department of Medicine, 2Department of Surgery, 3Nephrology Research and Training Center, and 4Comprehensive Cardiovascular Center, University of Alabama at Birmingham, Birmingham, Alabama, USA. 5Department of Veterans Affairs, Birmingham, Alabama, USA. 6Department of Anesthesiology and 7Department of Pulmonary, Allergy and Critical Care, Duke University School of Medicine, Durham, North Carolina, USA. The cardioprotective inducible enzyme heme oxygenase-1 (HO-1) degrades prooxidant heme into equimolar quantities of carbon monoxide, biliverdin, and iron. We hypothesized that HO-1 mediates cardiac protection, at least in part, by regulating mitochondrial quality control. We treated WT and HO-1 transgenic mice with the known mitochondrial toxin, doxorubicin (DOX). Relative to WT mice, mice globally overexpressing human HO-1 were protected from DOX-induced dilated cardiomyopathy, cardiac cytoarchitectural derangement, and infiltration of CD11b+ mononuclear phagocytes. Cardiac-specific overexpression of HO-1 ameliorated DOX-mediated dilation of the sarcoplasmic reticulum as well as mitochondrial disorganization in the form of mitochondrial fragmentation and increased numbers of damaged mitochondria in autophagic vacuoles. HO-1 overexpression promotes mitochondrial biogenesis by upregulating protein expression of NRF1, PGC1α, and TFAM, which was inhibited in WT animals treated with DOX. Concomitantly, HO-1 overexpression inhibited the upregulation of the mitochondrial fission mediator Fis1 and resulted in increased expression of the fusion mediators, Mfn1 and Mfn2. -

CDH12 Cadherin 12, Type 2 N-Cadherin 2 RPL5 Ribosomal
5 6 6 5 . 4 2 1 1 1 2 4 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 A A A A A A A A A A A A A A A A A A A A C C C C C C C C C C C C C C C C C C C C R R R R R R R R R R R R R R R R R R R R B , B B B B B B B B B B B B B B B B B B B , 9 , , , , 4 , , 3 0 , , , , , , , , 6 2 , , 5 , 0 8 6 4 , 7 5 7 0 2 8 9 1 3 3 3 1 1 7 5 0 4 1 4 0 7 1 0 2 0 6 7 8 0 2 5 7 8 0 3 8 5 4 9 0 1 0 8 8 3 5 6 7 4 7 9 5 2 1 1 8 2 2 1 7 9 6 2 1 7 1 1 0 4 5 3 5 8 9 1 0 0 4 2 5 0 8 1 4 1 6 9 0 0 6 3 6 9 1 0 9 0 3 8 1 3 5 6 3 6 0 4 2 6 1 0 1 2 1 9 9 7 9 5 7 1 5 8 9 8 8 2 1 9 9 1 1 1 9 6 9 8 9 7 8 4 5 8 8 6 4 8 1 1 2 8 6 2 7 9 8 3 5 4 3 2 1 7 9 5 3 1 3 2 1 2 9 5 1 1 1 1 1 1 5 9 5 3 2 6 3 4 1 3 1 1 4 1 4 1 7 1 3 4 3 2 7 6 4 2 7 2 1 2 1 5 1 6 3 5 6 1 3 6 4 7 1 6 5 1 1 4 1 6 1 7 6 4 7 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m -
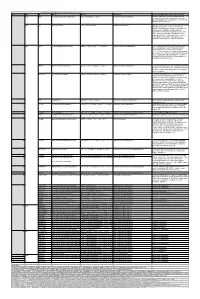
When the Reaction Is
Table S3. iJL1678-ME model modification (blocked reactions) Iter. Cat. ID Name Formula Subsystem Comments (When the reaction is turned on) 1 bp2 EDD 6-phosphogluconate dehydratase 6pgc_c⇌2ddg6p_c + h2o_c Pentose Phosphate Pathway Create a major effect of steep acetate overflow elevation in high growth. Comparing to the main glycolytic pathway, it is metabolicly less efficient but proteomicly more efficient. bp1 ICL Isocitrate lyase icit_c→glx_c + succ_c Anaplerotic Reactions Bypass for the main TCA cycle pathways from turning isocitrate to succinate, when ICL is turned on, Isocitrate dehydrogenase(ICDHyr), 2-Oxogluterate dehydrogenase(AKGDH) and Succinyl-CoA synthetase (ATP-forming,SUCOAS) would reduce. Ref. (1) and (2) shows that this reaction is off in higher growth. Ref. (3) shows that this reaction is converging to being off when the dynamic of respiration using enzyme kinetics is simulated. 2 bp1 ABTA 4-aminobutyrate transaminase 4abut_c + akg_c⇌glu__L_c + sucsal_c Arginine and Proline Metabolism Another backup pathway of succinate production, from 2-Oxoglutarate (akg). Respiration would be induced when it is on, since the flux through ETC(CYTBO3_4pp and ATPS4rpp) would increase. As it requires the co-factor pyridoxal 5'-phosphate(2−) to get catalyzed(4), indicating that this reaction is regulated by the flux of other reactions(pyridoxal 5'- phosphate(2-) production, etc.). 3 GLYAT Glycine C-acetyltransferase accoa_c + gly_c⇌2aobut_c + coa_c Glycine and Serine Metabolism A reaction that back up for the respiration. Reactions fluxes in TCA cycle would drop when this reaction is turned on. It also requires pyridoxal 5'-phosphate(2−) for the regulation. 4 NADTRHD NAD transhydrogenase nad_c + nadph_c⇌nadh_c + nadp_c Oxidative Phosphorylation A reaction that would make the transition between NAD and NADP metabolically more efficient. -
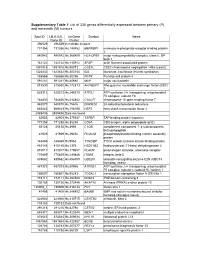
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

33 34 35 Lipid Synthesis Laptop
BI/CH 422/622 Liver cytosol ANABOLISM OUTLINE: Photosynthesis Carbohydrate Biosynthesis in Animals Biosynthesis of Fatty Acids and Lipids Fatty Acids Triacylglycerides contrasts Membrane lipids location & transport Glycerophospholipids Synthesis Sphingolipids acetyl-CoA carboxylase Isoprene lipids: fatty acid synthase Ketone Bodies ACP priming 4 steps Cholesterol Control of fatty acid metabolism isoprene synth. ACC Joining Reciprocal control of b-ox Cholesterol Synth. Diversification of fatty acids Fates Eicosanoids Cholesterol esters Bile acids Prostaglandins,Thromboxanes, Steroid Hormones and Leukotrienes Metabolism & transport Control ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Synthesis Degradation Ketone body Utilization Isoprene Biosynthesis 1 Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates are transferred stepwise from ATP to mevalonate. • A third phosphate from ATP is added at the hydroxyl, followed by decarboxylation and elimination catalyzed by pyrophospho- mevalonate decarboxylase creates a pyrophosphorylated 5-C product: D3-isopentyl pyrophosphate (IPP) (isoprene). • Isomerization to a second isoprene dimethylallylpyrophosphate (DMAPP) gives two activated isoprene IPP compounds that act as precursors for D3-isopentyl pyrophosphate Isopentyl-D-pyrophosphate all of the other lipids in this class isomerase DMAPP Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates -

The Human Flavoproteome
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Archives of Biochemistry and Biophysics 535 (2013) 150–162 Contents lists available at SciVerse ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi Review The human flavoproteome ⇑ Wolf-Dieter Lienhart, Venugopal Gudipati, Peter Macheroux Graz University of Technology, Institute of Biochemistry, Petersgasse 12, A-8010 Graz, Austria article info abstract Article history: Vitamin B2 (riboflavin) is an essential dietary compound used for the enzymatic biosynthesis of FMN and Received 17 December 2012 FAD. The human genome contains 90 genes encoding for flavin-dependent proteins, six for riboflavin and in revised form 21 February 2013 uptake and transformation into the active coenzymes FMN and FAD as well as two for the reduction to Available online 15 March 2013 the dihydroflavin form. Flavoproteins utilize either FMN (16%) or FAD (84%) while five human flavoen- zymes have a requirement for both FMN and FAD. The majority of flavin-dependent enzymes catalyze Keywords: oxidation–reduction processes in primary metabolic pathways such as the citric acid cycle, b-oxidation Coenzyme A and degradation of amino acids. Ten flavoproteins occur as isozymes and assume special functions in Coenzyme Q the human organism. Two thirds of flavin-dependent proteins are associated with disorders caused by Folate Heme allelic variants affecting protein function. Flavin-dependent proteins also play an important role in the Pyridoxal 50-phosphate biosynthesis of other essential cofactors and hormones such as coenzyme A, coenzyme Q, heme, pyri- Steroids doxal 50-phosphate, steroids and thyroxine. Moreover, they are important for the regulation of folate Thyroxine metabolites by using tetrahydrofolate as cosubstrate in choline degradation, reduction of N-5.10-meth- Vitamins ylenetetrahydrofolate to N-5-methyltetrahydrofolate and maintenance of the catalytically competent form of methionine synthase. -

(12) Patent Application Publication (10) Pub. No.: US 2017/0137846A1 ATSUMI Et Al
US 2017.0137846A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0137846A1 ATSUMI et al. (43) Pub. Date: May 18, 2017 (54) BACTERIA ENGINEERED FOR Related U.S. Application Data CONVERSION OF ETHYLENE TO (60) Provisional application No. 62/009,857, filed on Jun. N-BUTANOL 9, 2014. (71) Applicant: The Regents of the University of California, Oakland, CA (US) Publication Classification (51) Int. Cl. (72) Inventors: Shota ATSUMI, Davis, CA (US); CI2P 7/06 (2006.01) Michael D. TONEY, Davis, CA (US); CI2P 7/16 (2006.01) Gabriel M. RODRIGUEZ, Davis, CA CI2N 15/52 (2006.01) (US); Yohei TASHIRO, Davis, CA (52) U.S. Cl. (US); Justin B. SIEGEL, Davis, CA CPC ................ CI2P 7/06 (2013.01): CI2N 15/52 (US); D. Alexander CARLIN, Davis, (2013.01); C12Y402/01053 (2013.01); C12Y CA (US); Irina KORYAKINA, Davis, 402/01095 (2013.01); C12Y 114/13 (2013.01); CA (US); Shuchi H. DESAI, Davis, CI2Y 102/01002 (2013.01); C12Y 114/13069 CA (US) (2013.01); C12Y 203/01009 (2013.01); C12Y 101/01157 (2013.01); C12Y402/01 (2013.01); (73) Assignee: The Regents of the University of CI2Y 103/01038 (2013.01); C12Y 102/01003 California, Oakland, CA (US) (2013.01); C12P 7/16 (2013.01) (21) Appl. No.: 15/317,656 (57) ABSTRACT (22) PCT Fed: Jun. 9, 2015 The present disclosure provides recombinant bacteria with elevated production of ethanol and/or n-butanol from eth (86) PCT No.: PCT/US 15/34942 ylene. Methods for the production of the recombinant bac S 371 (c)(1), teria, as well as for use thereof for production of ethanol (2) Date: Dec.