CYP2A6) by P53
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amplitaq and Amplitaq Gold DNA Polymerase
AmpliTaq and AmpliTaq Gold DNA Polymerase The Most Referenced Brand of DNA Polymerase in the World Date: 2005-05 Notes: Authors are listed alphabetically J. Biol. Chem. (223) Ezoe, S., I. Matsumura, et al. (2005). "GATA Transcription Factors Inhibit Cytokine-dependent Growth and Survival of a Hematopoietic Cell Line through the Inhibition of STAT3 Activity." J. Biol. Chem. 280(13): 13163-13170. http://www.jbc.org/cgi/content/abstract/280/13/13163 Although GATA-1 and GATA-2 were shown to be essential for the development of hematopoietic cells by gene targeting experiments, they were also reported to inhibit the growth of hematopoietic cells. Therefore, in this study, we examined the effects of GATA-1 and GATA-2 on cytokine signals. A tamoxifen-inducible form of GATA-1 (GATA-1/ERT) showed a minor inhibitory effect on interleukin-3 (IL-3)-dependent growth of an IL-3-dependent cell line Ba/F3. On the other hand, it drastically inhibited TPO-dependent growth and gp130-mediated growth/survival of Ba/F3. Similarly, an estradiol-inducible form of GATA-2 (GATA-2/ER) disrupted thrombopoietin (TPO)-dependent growth and gp130-mediated growth/survival of Ba/F3. As for this mechanism, we found that both GATA-1 and GATA-2 directly bound to STAT3 both in vitro and in vivo and inhibited its DNA-binding activity in gel shift assays and chromatin immunoprecipitation assays, whereas they hardly affected STAT5 activity. In addition, endogenous GATA-1 was found to interact with STAT3 in normal megakaryocytes, suggesting that GATA-1 may inhibit STAT3 activity in normal hematopoietic cells. -

Characterisation of Bilirubin Metabolic Pathway in Hepatic Mitochondria Siti Nur Fadzilah Muhsain M.Sc
Characterisation of Bilirubin Metabolic Pathway in Hepatic Mitochondria Siti Nur Fadzilah Muhsain M.Sc. (Medical Research) 2005 Universiti Sains Malaysia Postgrad. Dip. (Toxicology) 2003 University of Surrey B.Sc.(Biomed. Sc.) 2000 Universiti Putra Malaysia A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2014 School of Medicine ABSTRACT Bilirubin (BR), a toxic waste product of degraded haem, is a potent antioxidant at physiological concentrations. To achieve the maximum benefit of BR, its intracellular level needs to be carefully regulated. A system comprising of two enzymes, haem oxygenase-1 (HMOX1) and cytochrome P450 2A5 (CYP2A5) exists in the endoplasmic reticulum (ER), responsible for regulating BR homeostasis. This system is induced in response to oxidative stress. In this thesis, oxidative stress caused accumulation of these enzymes in mitochondria — major producers and targets of reactive oxygen species (ROS) — is demonstrated. To understand the significance of this intracellular targeting, properties of microsomal and mitochondrial BR metabolising enzymes were compared and the capacity of mitochondrial CYP2A5 to oxidise BR in response to oxidative stress is reported. Microsomes and mitochondrial fractions were isolated from liver homogenates of DBA/2J mice, administered with sub-toxic dose of pyrazole, an oxidant stressor. The purity of extracted organelles was determined by analysing the expressions and activities of their respective marker enzymes. HMOX1 and CYP2A5 were significantly increased in microsomes and even more so in mitochondria in response to pyrazole-induced oxidative stress. By contrast, the treatment did not increase either microsomes or mitochondrial Uridine-diphosphate-glucuronosyltransferase 1A1 (UGT1A1), the sole enzyme that catalyses BR elimination through glucuronidation. -

Heme Oxygenase-1 Regulates Mitochondrial Quality Control in the Heart
RESEARCH ARTICLE Heme oxygenase-1 regulates mitochondrial quality control in the heart Travis D. Hull,1,2 Ravindra Boddu,1,3 Lingling Guo,2,3 Cornelia C. Tisher,1,3 Amie M. Traylor,1,3 Bindiya Patel,1,4 Reny Joseph,1,3 Sumanth D. Prabhu,1,4,5 Hagir B. Suliman,6 Claude A. Piantadosi,7 Anupam Agarwal,1,3,5 and James F. George2,3,4 1Department of Medicine, 2Department of Surgery, 3Nephrology Research and Training Center, and 4Comprehensive Cardiovascular Center, University of Alabama at Birmingham, Birmingham, Alabama, USA. 5Department of Veterans Affairs, Birmingham, Alabama, USA. 6Department of Anesthesiology and 7Department of Pulmonary, Allergy and Critical Care, Duke University School of Medicine, Durham, North Carolina, USA. The cardioprotective inducible enzyme heme oxygenase-1 (HO-1) degrades prooxidant heme into equimolar quantities of carbon monoxide, biliverdin, and iron. We hypothesized that HO-1 mediates cardiac protection, at least in part, by regulating mitochondrial quality control. We treated WT and HO-1 transgenic mice with the known mitochondrial toxin, doxorubicin (DOX). Relative to WT mice, mice globally overexpressing human HO-1 were protected from DOX-induced dilated cardiomyopathy, cardiac cytoarchitectural derangement, and infiltration of CD11b+ mononuclear phagocytes. Cardiac-specific overexpression of HO-1 ameliorated DOX-mediated dilation of the sarcoplasmic reticulum as well as mitochondrial disorganization in the form of mitochondrial fragmentation and increased numbers of damaged mitochondria in autophagic vacuoles. HO-1 overexpression promotes mitochondrial biogenesis by upregulating protein expression of NRF1, PGC1α, and TFAM, which was inhibited in WT animals treated with DOX. Concomitantly, HO-1 overexpression inhibited the upregulation of the mitochondrial fission mediator Fis1 and resulted in increased expression of the fusion mediators, Mfn1 and Mfn2. -

CDH12 Cadherin 12, Type 2 N-Cadherin 2 RPL5 Ribosomal
5 6 6 5 . 4 2 1 1 1 2 4 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 A A A A A A A A A A A A A A A A A A A A C C C C C C C C C C C C C C C C C C C C R R R R R R R R R R R R R R R R R R R R B , B B B B B B B B B B B B B B B B B B B , 9 , , , , 4 , , 3 0 , , , , , , , , 6 2 , , 5 , 0 8 6 4 , 7 5 7 0 2 8 9 1 3 3 3 1 1 7 5 0 4 1 4 0 7 1 0 2 0 6 7 8 0 2 5 7 8 0 3 8 5 4 9 0 1 0 8 8 3 5 6 7 4 7 9 5 2 1 1 8 2 2 1 7 9 6 2 1 7 1 1 0 4 5 3 5 8 9 1 0 0 4 2 5 0 8 1 4 1 6 9 0 0 6 3 6 9 1 0 9 0 3 8 1 3 5 6 3 6 0 4 2 6 1 0 1 2 1 9 9 7 9 5 7 1 5 8 9 8 8 2 1 9 9 1 1 1 9 6 9 8 9 7 8 4 5 8 8 6 4 8 1 1 2 8 6 2 7 9 8 3 5 4 3 2 1 7 9 5 3 1 3 2 1 2 9 5 1 1 1 1 1 1 5 9 5 3 2 6 3 4 1 3 1 1 4 1 4 1 7 1 3 4 3 2 7 6 4 2 7 2 1 2 1 5 1 6 3 5 6 1 3 6 4 7 1 6 5 1 1 4 1 6 1 7 6 4 7 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m -

Dissertation
Dissertation Submitted to the Combined Faculties for the Natural Sciences and for Mathematics of the Ruperto-Carola University of Heidelberg, Germany for the Degree of Doctor of Natural Sciences Presented by Ann-Cathrin Hofer (M.Sc.) Born in Heidelberg, Germany Oral Examination: 12th of September 2016 Regulatory T cells protect the neonatal liver and secure the hepatic circadian rhythm Referees 1st Referee: Prof. Dr. Peter Angel 2nd Referee: Dr. Markus Feuerer This dissertation was performed and written during the period from November 2012 to May 2016 in the German Cancer Research Center (DKFZ) under the supervision of Prof. Dr. Peter Angel and direct supervision of Dr. Markus Feuerer. The dissertation was submitted to the Combined Faculties for the Natural Sciences and for Mathematics of the Ruperto-Carola University of Heidelberg, Germany in June 2016. German Cancer Research Center (DKFZ) Immune Tolerance (D100) Im Neuenheimer Feld 280 69120 Heidelberg, Germany I II Confirmation Hereby, I confirm that I have written this thesis independently, using only the results of my investigation unless otherwise stated. Furthermore, I declare that I have not submitted this thesis for a degree to any other academic or similar institution. Parts of this dissertation have been submitted for publishing: Regulatory T cells protect the liver early in life and safeguard the hepatic circadian rhythm Ann-Cathrin Hofer, Thomas Hielscher, David M. Richards, Michael Delacher, Ulrike Träger, Sophia Föhr, Artyom Vlasov, Marvin Wäsch, Marieke Esser, Annette Kopp-Schneider, Achim Breiling, Frank Lyko, Ursula Klingmüller, Peter Angel, Jakub Abramson, Jeroen Krijgsveld & Markus Feuerer Parts of the experiments in this dissertation were performed in collaboration with other research groups as follows: CG methylation analysis with the 454 pyrosequencing technology: Division of Epigenetics, DKFZ, Heidelberg Dr. -
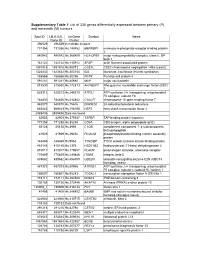
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Solarbio Catalogue with PRICES
CAS Name Grade Purity Biochemical Reagent Biochemical Reagent 75621-03-3 C8390-1 3-((3-Cholamidopropyl)dimethylammonium)-1-propanesulfonateCHAPS Ultra Pure Grade 1g 75621-03-3 C8390-5 3-((3-Cholamidopropyl)dimethylammonium)-1-propanesulfonateCHAPS 5g 57-09-0 C8440-25 Cetyl-trimethyl Ammonium Bromide CTAB High Pure Grade ≥99.0% 25g 57-09-0 C8440-100 Cetyl-trimethyl Ammonium Bromide CTAB High Pure Grade ≥99.0% 100g 57-09-0 C8440-500 Cetyl-trimethyl Ammonium Bromide CTAB High Pure Grade ≥99.0% 500g E1170-100 0.5M EDTA (PH8.0) 100ml E1170-500 0.5M EDTA (PH8.0) 500ml 6381-92-6 E8030-100 EDTA disodium salt dihydrate EDTA Na2 Biotechnology Grade ≥99.0% 100g 6381-92-6 E8030-500 EDTA disodium salt dihydrate EDTA Na2 Biotechnology Grade ≥99.0% 500g 6381-92-6 E8030-1000 EDTA disodium salt dihydrate EDTA Na2 Biotechnology Grade ≥99.0% 1kg 6381-92-6 E8030-5000 EDTA disodium salt dihydrate EDTA Na2 Biotechnology Grade ≥99.0% 5kg 60-00-4 E8040-100 Ethylenediaminetetraacetic acid EDTA Ultra Pure Grade ≥99.5% 100g 60-00-4 E8040-500 Ethylenediaminetetraacetic acid EDTA Ultra Pure Grade ≥99.5% 500g 60-00-4 E8040-1000 Ethylenediaminetetraacetic acid EDTA Ultra Pure Grade ≥99.5% 1kg 67-42-5 E8050-5 Ethylene glycol-bis(2-aminoethylether)-N,N,NEGTA′,N′-tetraacetic acid Ultra Pure Grade ≥97.0% 5g 67-42-5 E8050-10 Ethylene glycol-bis(2-aminoethylether)-N,N,NEGTA′,N′-tetraacetic acid Ultra Pure Grade ≥97.0% 10g 50-01-1 G8070-100 Guanidine Hydrochloride Guanidine HCl ≥98.0%(AT) 100g 50-01-1 G8070-500 Guanidine Hydrochloride Guanidine HCl ≥98.0%(AT) 500g 56-81-5 -

1 Molecular Physiology and Pathophysiology of Bilirubin Handling by the Blood, Liver
1 1 MOLECULAR PHYSIOLOGY AND PATHOPHYSIOLOGY OF BILIRUBIN HANDLING BY THE BLOOD, LIVER, 2 INTESTINE, AND BRAIN IN THE NEWBORN 3 THOR W.R. HANSEN1, RONALD J. WONG2, DAVID K. STEVENSON2 4 1Division of Paediatric and Adolescent Medicine, Institute of Clinical Medicine, Faculty of Medicine, 5 University of Oslo, Norway 6 2Department of Pediatrics, Stanford University School of Medicine, Stanford CA, USA 7 __________________________________________________________________________________ 8 2 9 I. Introduction 10 II. Bilirubin in the Body 11 A. Bilirubin Chemistry 12 1. Bilirubin structure 13 2. Bilirubin solubility 14 3. Bilirubin isomers 15 4. Heme degradation 16 5. Biliverdin and biliverdin reductase (BVR) 17 B. Bilirubin as an Antioxidant 18 C. Bilirubin as a Toxin 19 1. Bilirubin effects on enzyme activity 20 2. Toxicity of bilirubin conjugates and isomers 21 D. Other Functions/Roles 22 1. Drug displacement by bilirubin 23 2. Bilirubin interactions with the immune system and 24 inflammatory/infectious mechanisms 25 III. The Production of Bilirubin in the Body 26 A. Heme Catabolism and Its Regulation 27 1. Genetic variants in bilirubin production 28 B. The Effect of Hemolysis 29 1. Disorders associated with increased bilirubin production 30 IV. Bilirubin Binding and Transport in Blood 31 V. Bilirubin in the Liver 32 A. Hepatocellular Uptake and Intracellular Processing 33 B. Bilirubin Conjugation 34 1. Genetic variants in bilirubin conjugation 3 35 a. Crigler-Najjar syndrome type I 36 b. Crigler-Najjar syndrome type II 37 c. Gilbert syndrome 38 2. Genetic variants in transporter proteins 39 C. Bilirubin Excretion 40 VI. Bilirubin in the Intestines 41 A. -

Biocatalyzed Synthesis of Statins: a Sustainable Strategy for the Preparation of Valuable Drugs
catalysts Review Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs Pilar Hoyos 1, Vittorio Pace 2 and Andrés R. Alcántara 1,* 1 Department of Chemistry in Pharmaceutical Sciences, Faculty of Pharmacy, Complutense University of Madrid, Campus de Moncloa, E-28040 Madrid, Spain; [email protected] 2 Department of Pharmaceutical Chemistry, Faculty of Life Sciences, Althanstrasse 14, A-1090 Vienna, Austria; [email protected] * Correspondence: [email protected]; Tel.: +34-91-394-1823 Received: 25 February 2019; Accepted: 9 March 2019; Published: 14 March 2019 Abstract: Statins, inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, are the largest selling class of drugs prescribed for the pharmacological treatment of hypercholesterolemia and dyslipidaemia. Statins also possess other therapeutic effects, called pleiotropic, because the blockade of the conversion of HMG-CoA to (R)-mevalonate produces a concomitant inhibition of the biosynthesis of numerous isoprenoid metabolites (e.g., geranylgeranyl pyrophosphate (GGPP) or farnesyl pyrophosphate (FPP)). Thus, the prenylation of several cell signalling proteins (small GTPase family members: Ras, Rac, and Rho) is hampered, so that these molecular switches, controlling multiple pathways and cell functions (maintenance of cell shape, motility, factor secretion, differentiation, and proliferation) are regulated, leading to beneficial effects in cardiovascular health, regulation of the immune system, anti-inflammatory and immunosuppressive properties, prevention and treatment of sepsis, treatment of autoimmune diseases, osteoporosis, kidney and neurological disorders, or even in cancer therapy. Thus, there is a growing interest in developing more sustainable protocols for preparation of statins, and the introduction of biocatalyzed steps into the synthetic pathways is highly advantageous—synthetic routes are conducted under mild reaction conditions, at ambient temperature, and can use water as a reaction medium in many cases. -

Identification of Systemic Immune Response Markers Through
Gray et al. Veterinary Research (2015) 46:7 DOI 10.1186/s13567-014-0138-z VETERINARY RESEARCH RESEARCH Open Access Identification of systemic immune response markers through metabolomic profiling of plasma from calves given an intra-nasally delivered respiratory vaccine Darren W Gray1*, Michael D Welsh2, Simon Doherty2, Fawad Mansoor2, Olivier P Chevallier1, Christopher T Elliott1 and Mark H Mooney1 Abstract Vaccination procedures within the cattle industry are important disease control tools to minimize economic and welfare burdens associated with respiratory pathogens. However, new vaccine, antigen and carrier technologies are required to combat emerging viral strains and enhance the efficacy of respiratory vaccines, particularly at the point of pathogen entry. New technologies, specifically metabolomic profiling, could be applied to identify metabolite immune-correlates representative of immune protection following vaccination aiding in the design and screening of vaccine candidates. This study for the first time demonstrates the ability of untargeted UPLC-MS metabolomic profiling to identify metabolite immune correlates characteristic of immune responses following mucosal vaccination in calves. Male Holstein Friesian calves were vaccinated with Pfizer Rispoval® PI3 + RSV intranasal vaccine and metabolomic profiling of post-vaccination plasma revealed 12 metabolites whose peak intensities differed significantly from controls. Plasma levels of glycocholic acid, N-[(3α,5β,12α)-3,12-Dihydroxy-7,24-dioxocholan-24-yl]glycine, uric acid and -

Biliverdin Reductase: a Major Physiologic Cytoprotectant
Biliverdin reductase: A major physiologic cytoprotectant David E. Baran˜ ano*, Mahil Rao*, Christopher D. Ferris†, and Solomon H. Snyder*‡§¶ Departments of *Neuroscience, ‡Pharmacology and Molecular Sciences, and §Psychiatry and Behavioral Sciences, The Johns Hopkins University School of Medicine, Baltimore, MD 21205; and †Department of Medicine, Division of Gastroenterology, C-2104 Medical Center North, Vanderbilt University Medical Center, Nashville, TN 37232-2279 Contributed by Solomon H. Snyder, October 16, 2002 Bilirubin, an abundant pigment that causes jaundice, has long hypothesize that bilirubin acts in a catalytic fashion whereby lacked any clear physiologic role. It arises from enzymatic reduction bilirubin oxidized to biliverdin is rapidly reduced back to bili- by biliverdin reductase of biliverdin, a product of heme oxygenase rubin, a process that could readily afford 10,000-fold amplifica- activity. Bilirubin is a potent antioxidant that we show can protect tion (13). Here we establish that a redox cycle based on BVRA cells from a 10,000-fold excess of H2O2. We report that bilirubin is activity provides physiologic cytoprotection as BVRA depletion a major physiologic antioxidant cytoprotectant. Thus, cellular de- exacerbates the formation of reactive oxygen species (ROS) and pletion of bilirubin by RNA interference markedly augments tissue augments cell death. levels of reactive oxygen species and causes apoptotic cell death. Depletion of glutathione, generally regarded as a physiologic Methods antioxidant cytoprotectant, elicits lesser increases in reactive ox- All chemicals were obtained from Sigma unless otherwise ygen species and cell death. The potent physiologic antioxidant indicated. actions of bilirubin reflect an amplification cycle whereby bilirubin, acting as an antioxidant, is itself oxidized to biliverdin and then Cell Culture and Viability Measurements. -

The Extent of Intracellular Accumulation of Bilirubin Determines Its Anti- Or Pro-Oxidant Effect
International Journal of Molecular Sciences Article The Extent of Intracellular Accumulation of Bilirubin Determines Its Anti- or Pro-Oxidant Effect Annalisa Bianco 1,2, Aleš Dvoˇrák 3 , Nikola Capková 3, Camille Gironde 4, Claudio Tiribelli 1 , Christophe Furger 4, Libor Vitek 3,5 and Cristina Bellarosa 1,* 1 Italian Liver Foundation (FIF), Bldg Q—AREA Science Park Basovizza, SS14 Km 163,5, 34149 Trieste, Italy; [email protected] (A.B.); [email protected] (C.T.) 2 Department of Life Sciences, University of Trieste, 34127 Trieste, Italy 3 Institute of Medical Biochemistry and Laboratory Diagnostics, Faculty General Hospital and 1st Faculty of Medicine, Charles University, 121 08 Prague 2, Czech Republic; [email protected] (A.D.); [email protected] (N.C.); [email protected] (L.V.) 4 AOP/MH2F Team, LAAS-CNRS, 7 avenue de l’Europe, 31400 Toulouse, France; [email protected] (C.G.); [email protected] (C.F.) 5 4th Department of Internal Medicine, Faculty General Hospital and 1st Faculty of Medicine, Charles University, 121 08 Prague 2, Czech Republic * Correspondence: [email protected] Received: 24 September 2020; Accepted: 28 October 2020; Published: 30 October 2020 Abstract: Background: Severe hyperbilirubinemia can cause permanent neurological damage in particular in neonates, whereas mildly elevated serum bilirubin protects from various oxidative stress-mediated diseases. The present work aimed to establish the intracellular unconjugated bilirubin concentrations (iUCB) thresholds differentiating between anti- and pro-oxidant effects. Methods: Hepatic (HepG2), heart endothelial (H5V), kidney tubular (HK2) and neuronal (SH-SY5Y) cell lines were exposed to increasing concentration of bilirubin. iUCB, cytotoxicity, intracellular reactive oxygen species (ROS) concentrations, and antioxidant capacity (50% efficacy concentration (EC50)) were determined.