Heme Oxygenase-1 Regulates Mitochondrial Quality Control in the Heart
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amplitaq and Amplitaq Gold DNA Polymerase
AmpliTaq and AmpliTaq Gold DNA Polymerase The Most Referenced Brand of DNA Polymerase in the World Date: 2005-05 Notes: Authors are listed alphabetically J. Biol. Chem. (223) Ezoe, S., I. Matsumura, et al. (2005). "GATA Transcription Factors Inhibit Cytokine-dependent Growth and Survival of a Hematopoietic Cell Line through the Inhibition of STAT3 Activity." J. Biol. Chem. 280(13): 13163-13170. http://www.jbc.org/cgi/content/abstract/280/13/13163 Although GATA-1 and GATA-2 were shown to be essential for the development of hematopoietic cells by gene targeting experiments, they were also reported to inhibit the growth of hematopoietic cells. Therefore, in this study, we examined the effects of GATA-1 and GATA-2 on cytokine signals. A tamoxifen-inducible form of GATA-1 (GATA-1/ERT) showed a minor inhibitory effect on interleukin-3 (IL-3)-dependent growth of an IL-3-dependent cell line Ba/F3. On the other hand, it drastically inhibited TPO-dependent growth and gp130-mediated growth/survival of Ba/F3. Similarly, an estradiol-inducible form of GATA-2 (GATA-2/ER) disrupted thrombopoietin (TPO)-dependent growth and gp130-mediated growth/survival of Ba/F3. As for this mechanism, we found that both GATA-1 and GATA-2 directly bound to STAT3 both in vitro and in vivo and inhibited its DNA-binding activity in gel shift assays and chromatin immunoprecipitation assays, whereas they hardly affected STAT5 activity. In addition, endogenous GATA-1 was found to interact with STAT3 in normal megakaryocytes, suggesting that GATA-1 may inhibit STAT3 activity in normal hematopoietic cells. -

CYP2A6) by P53
Transcriptional Regulation of Human Stress Responsive Cytochrome P450 2A6 (CYP2A6) by p53 Hao Hu M.Biotech. (Biotechnology) 2012 The University of Queensland B.B.A. 2009 University of Electronic Science and Technology of China B.Sc. (Pharmacy) 2009 University of Electronic Science and Technology of China A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2016 School of Medicine ABSTRACT Human cytochrome P450 (CYP) 2A6 is highly expressed in the liver and the encoding gene is regulated by various stress activated transcription factors, such as the nuclear factor (erythroid-derived 2)-like 2 (Nrf-2). Unlike the other xenobiotic metabolising CYP enzymes (XMEs), CYP2A6 only plays a minor role in xenobiotic metabolism. The CYP2A6 is highly induced by multiple forms of cellular stress conditions, where XMEs expression is normally inhibited. Recent findings suggest that the CYP2A6 plays an important role in regulating BR homeostasis. A computer based sequence analysis on the 3 kb proximate CYP2A6 promoter revealed several putative binding sites for p53, a protein that mediates regulation of antioxidant and apoptosis pathways. In this study, the role of p53 in CYP2A6 gene regulation is demonstrated. The site closest to transcription start site (TSS) is highly homologous with the p53 consensus sequence. The p53 responsiveness of this site was confirmed by transfections with various stepwise deleted of CYP2A6-5’-Luc constructs containing the putative p53RE. Deletion of the putative p53RE resulted in a total abolishment of p53 responsiveness of CYP2A6 promoter. Specific binding of p53 to the putative p53RE was detected by electrophoresis mobility shift assay. -

Characterisation of Bilirubin Metabolic Pathway in Hepatic Mitochondria Siti Nur Fadzilah Muhsain M.Sc
Characterisation of Bilirubin Metabolic Pathway in Hepatic Mitochondria Siti Nur Fadzilah Muhsain M.Sc. (Medical Research) 2005 Universiti Sains Malaysia Postgrad. Dip. (Toxicology) 2003 University of Surrey B.Sc.(Biomed. Sc.) 2000 Universiti Putra Malaysia A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2014 School of Medicine ABSTRACT Bilirubin (BR), a toxic waste product of degraded haem, is a potent antioxidant at physiological concentrations. To achieve the maximum benefit of BR, its intracellular level needs to be carefully regulated. A system comprising of two enzymes, haem oxygenase-1 (HMOX1) and cytochrome P450 2A5 (CYP2A5) exists in the endoplasmic reticulum (ER), responsible for regulating BR homeostasis. This system is induced in response to oxidative stress. In this thesis, oxidative stress caused accumulation of these enzymes in mitochondria — major producers and targets of reactive oxygen species (ROS) — is demonstrated. To understand the significance of this intracellular targeting, properties of microsomal and mitochondrial BR metabolising enzymes were compared and the capacity of mitochondrial CYP2A5 to oxidise BR in response to oxidative stress is reported. Microsomes and mitochondrial fractions were isolated from liver homogenates of DBA/2J mice, administered with sub-toxic dose of pyrazole, an oxidant stressor. The purity of extracted organelles was determined by analysing the expressions and activities of their respective marker enzymes. HMOX1 and CYP2A5 were significantly increased in microsomes and even more so in mitochondria in response to pyrazole-induced oxidative stress. By contrast, the treatment did not increase either microsomes or mitochondrial Uridine-diphosphate-glucuronosyltransferase 1A1 (UGT1A1), the sole enzyme that catalyses BR elimination through glucuronidation. -

CDH12 Cadherin 12, Type 2 N-Cadherin 2 RPL5 Ribosomal
5 6 6 5 . 4 2 1 1 1 2 4 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 A A A A A A A A A A A A A A A A A A A A C C C C C C C C C C C C C C C C C C C C R R R R R R R R R R R R R R R R R R R R B , B B B B B B B B B B B B B B B B B B B , 9 , , , , 4 , , 3 0 , , , , , , , , 6 2 , , 5 , 0 8 6 4 , 7 5 7 0 2 8 9 1 3 3 3 1 1 7 5 0 4 1 4 0 7 1 0 2 0 6 7 8 0 2 5 7 8 0 3 8 5 4 9 0 1 0 8 8 3 5 6 7 4 7 9 5 2 1 1 8 2 2 1 7 9 6 2 1 7 1 1 0 4 5 3 5 8 9 1 0 0 4 2 5 0 8 1 4 1 6 9 0 0 6 3 6 9 1 0 9 0 3 8 1 3 5 6 3 6 0 4 2 6 1 0 1 2 1 9 9 7 9 5 7 1 5 8 9 8 8 2 1 9 9 1 1 1 9 6 9 8 9 7 8 4 5 8 8 6 4 8 1 1 2 8 6 2 7 9 8 3 5 4 3 2 1 7 9 5 3 1 3 2 1 2 9 5 1 1 1 1 1 1 5 9 5 3 2 6 3 4 1 3 1 1 4 1 4 1 7 1 3 4 3 2 7 6 4 2 7 2 1 2 1 5 1 6 3 5 6 1 3 6 4 7 1 6 5 1 1 4 1 6 1 7 6 4 7 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m -
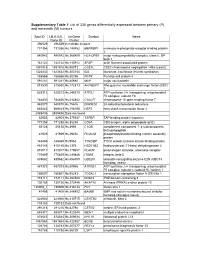
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Crystal Structure of Bacterial Succinate:Quinone Oxidoreductase Flavoprotein Sdha in Complex with Its Assembly Factor Sdhe
Crystal structure of bacterial succinate:quinone oxidoreductase flavoprotein SdhA in complex with its assembly factor SdhE Megan J. Mahera,1, Anuradha S. Heratha, Saumya R. Udagedaraa, David A. Dougana, and Kaye N. Truscotta,1 aDepartment of Biochemistry and Genetics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC 3086, Australia Edited by Amy C. Rosenzweig, Northwestern University, Evanston, IL, and approved February 14, 2018 (received for review January 4, 2018) Succinate:quinone oxidoreductase (SQR) functions in energy me- quinol:FRD), respectively (13, 15). The importance of this protein tabolism, coupling the tricarboxylic acid cycle and electron transport family, in normal cellular metabolism, is manifested by the iden- chain in bacteria and mitochondria. The biogenesis of flavinylated tification of a mutation in human SDHAF2 (Gly78Arg), which is SdhA, the catalytic subunit of SQR, is assisted by a highly conserved linked to an inherited neuroendocrine disorder, PGL2 (10). Cur- assembly factor termed SdhE in bacteria via an unknown mecha- rently, however, the role of SdhE in flavinylation remains poorly nism. By using X-ray crystallography, we have solved the structure understood. To date, three different modes of action for SdhE/ of Escherichia coli SdhE in complex with SdhA to 2.15-Å resolution. Sdh5 have been proposed, suggesting that SdhE facilitates the Our structure shows that SdhE makes a direct interaction with the binding and delivery of FAD (13), acts as a chaperone for SdhA flavin adenine dinucleotide-linked residue His45 in SdhA and main- (10), or catalyzes the attachment of FAD (10). Moreover, the re- tains the capping domain of SdhA in an “open” conformation. -

Updates on the Genetics of Neuroendocrine Tumors
Updates on the Genetics of Neuroendocrine Tumors NET Patient Conference March 8, 2019 Anna Raper, MS, CGC Division of Translational Medicine and Human Genetics No disclosures 2 3 Overview 1. Cancer/tumor genetics 2. Genetics of neuroendocrine tumors sciencemag.org 4 The Genetics of Cancers and Tumors Hereditary v. Familial v. Sporadic Germline v. somatic genetics Risk When to suspect hereditary susceptibility 5 Cancer Distribution - General Hereditary (5-10%) • Specific gene variant is inherited in family • Associated with increased tumor/cancer risk Familial (10-20%) • Multiple genes and environmental factors may be involved • Some increased tumor/cancer risk Sporadic • Occurs by chance, or related to environmental factors • General population tumor/cancer risk 6 What are genes again? 7 Normal gene Pathogenic gene variant (“mutation”) kintalk.org 8 Cancer is a genetic disease kintalk.org 9 Germline v. Somatic gene mutations 10 Hereditary susceptibility to cancer Germline mutations Depending on the gene, increased risk for certain tumor/cancer types Does not mean an individual WILL develop cancer, but could change screening and management recommendations National Cancer Institute 11 Features that raise suspicion for hereditary condition Specific tumor types Early ages of diagnosis compared to the general population Multiple or bilateral (affecting both sides) tumors Family history • Clustering of certain tumor types • Multiple generations affected • Multiple siblings affected 12 When is genetic testing offered? A hereditary -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Function and Structure of Complex Ii of The
Annu. Rev. Biochem. 2003. 72:77–109 doi: 10.1146/annurev.biochem.72.121801.161700 FUNCTION AND STRUCTURE OF COMPLEX II * OF THE RESPIRATORY CHAIN Gary Cecchini Molecular Biology Division, Veterans Administration Medical Center, San Francisco, California 94121, and Department of Biochemistry and Biophysics, University of California, San Francisco, California 94143; email: [email protected] Key Words succinate dehydrogenase, fumarate reductase, quinone oxidoreductase, reactive oxygen species, respiratory chain f Abstract Complex II is the only membrane-bound component of the Krebs cycle and in addition functions as a member of the electron transport chain in mitochondria and in many bacteria. A recent X-ray structural solution of members of the complex II family of proteins has provided important insights into their function. One feature of the complex II structures is a linear electron transport chain that extends from the flavin and iron-sulfur redox cofactors in the membrane extrinsic domain to the quinone and b heme cofactors in the membrane domain. Exciting recent developments in relation to disease in humans and the formation of reactive oxygen species by complex II point to its overall importance in cellular physiology. CONTENTS OVERVIEW OF MEMBRANE-BOUND RESPIRATORY CHAIN .......... 78 COMPLEX II .......................................... 80 OVERALL STRUCTURE OF COMPLEX II ....................... 83 FLAVOPROTEIN SUBUNIT AND FORMATION OF THE COVALENT FAD LINKAGE............................................ 87 CATALYSIS IN COMPLEX II................................ 91 IRON-SULFUR CLUSTERS OF COMPLEX II AND THE ELECTRON TRANS- FER PATHWAY........................................ 94 MEMBRANE DOMAIN OF COMPLEX II ........................ 95 ROLE OF THE b HEME IN COMPLEX II ........................ 99 RELATION OF COMPLEX II TO DISEASE AND REACTIVE OXYGEN SPECIES ............................................100 CONCLUDING REMARKS .................................104 *The U.S. -

First-Positive Surveillance Screening in an Asymptomatic SDHA Germline Mutation Carrier
ID: 19-0005 -19-0005 G White and others SDHA surveillance screen ID: 19-0005; May 2019 detected PGL DOI: 10.1530/EDM-19-0005 First-positive surveillance screening in an asymptomatic SDHA germline mutation carrier Correspondence should be addressed Gemma White, Nicola Tufton and Scott A Akker to S A Akker Email Department of Endocrinology, St. Bartholomew’s Hospital, Barts Health NHS Trust, London, UK [email protected] Summary At least 40% of phaeochromocytomas and paraganglioma’s (PPGLs) are associated with an underlying genetic mutation. The understanding of the genetic landscape of these tumours has rapidly evolved, with 18 associated genes now identified.Amongthese,mutationsinthesubunitsofsuccinatedehydrogenasecomplex(SDH) are the most common, causing around half of familial PPGL cases. Occurrence of PPGLs in carriers of SDHB, SDHC and SDHD subunit mutations has been long reported, but it is only recently that variants in the SDHA subunit have been linked to PPGL formation. Previously documented cases have, to our knowledge, only been found in isolated cases where pathogenic SDHA variants wereidentifiedretrospectively.Wereportthecaseofanasymptomaticsuspectedcarotidbodytumourfoundduring surveillance screening in a 72-year-old female who is a known carrier of a germline SDHA pathogenic variant. To our knowledge,thisisthefirstscreenthatdetectedPPGLfoundinapreviouslyidentifiedSDHA pathogenic variant carrier, duringsurveillanceimaging.Thisfindingsupportstheuseofcascadegenetictestingandsurveillancescreeninginall carriers of a pathogenic SDHA variant. Learning points: •• SDH mutations are important causes of PPGL disease. •• SDHA is much rarer compared to SDHB and SDHD mutations. •• Pathogenicity and penetrance are yet to be fully determined in cases of SDHA-related PPGL. •• Surveillance screening should be used for SDHA PPGL cases to identify recurrence, metastasis or metachronous disease. •• Surveillance screening for SDH-relateddiseaseshouldbeperformedinidentifiedcarriersofapathogenicSDHA variant. -

Biocatalyzed Synthesis of Statins: a Sustainable Strategy for the Preparation of Valuable Drugs
catalysts Review Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs Pilar Hoyos 1, Vittorio Pace 2 and Andrés R. Alcántara 1,* 1 Department of Chemistry in Pharmaceutical Sciences, Faculty of Pharmacy, Complutense University of Madrid, Campus de Moncloa, E-28040 Madrid, Spain; [email protected] 2 Department of Pharmaceutical Chemistry, Faculty of Life Sciences, Althanstrasse 14, A-1090 Vienna, Austria; [email protected] * Correspondence: [email protected]; Tel.: +34-91-394-1823 Received: 25 February 2019; Accepted: 9 March 2019; Published: 14 March 2019 Abstract: Statins, inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, are the largest selling class of drugs prescribed for the pharmacological treatment of hypercholesterolemia and dyslipidaemia. Statins also possess other therapeutic effects, called pleiotropic, because the blockade of the conversion of HMG-CoA to (R)-mevalonate produces a concomitant inhibition of the biosynthesis of numerous isoprenoid metabolites (e.g., geranylgeranyl pyrophosphate (GGPP) or farnesyl pyrophosphate (FPP)). Thus, the prenylation of several cell signalling proteins (small GTPase family members: Ras, Rac, and Rho) is hampered, so that these molecular switches, controlling multiple pathways and cell functions (maintenance of cell shape, motility, factor secretion, differentiation, and proliferation) are regulated, leading to beneficial effects in cardiovascular health, regulation of the immune system, anti-inflammatory and immunosuppressive properties, prevention and treatment of sepsis, treatment of autoimmune diseases, osteoporosis, kidney and neurological disorders, or even in cancer therapy. Thus, there is a growing interest in developing more sustainable protocols for preparation of statins, and the introduction of biocatalyzed steps into the synthetic pathways is highly advantageous—synthetic routes are conducted under mild reaction conditions, at ambient temperature, and can use water as a reaction medium in many cases. -

Identification of Systemic Immune Response Markers Through
Gray et al. Veterinary Research (2015) 46:7 DOI 10.1186/s13567-014-0138-z VETERINARY RESEARCH RESEARCH Open Access Identification of systemic immune response markers through metabolomic profiling of plasma from calves given an intra-nasally delivered respiratory vaccine Darren W Gray1*, Michael D Welsh2, Simon Doherty2, Fawad Mansoor2, Olivier P Chevallier1, Christopher T Elliott1 and Mark H Mooney1 Abstract Vaccination procedures within the cattle industry are important disease control tools to minimize economic and welfare burdens associated with respiratory pathogens. However, new vaccine, antigen and carrier technologies are required to combat emerging viral strains and enhance the efficacy of respiratory vaccines, particularly at the point of pathogen entry. New technologies, specifically metabolomic profiling, could be applied to identify metabolite immune-correlates representative of immune protection following vaccination aiding in the design and screening of vaccine candidates. This study for the first time demonstrates the ability of untargeted UPLC-MS metabolomic profiling to identify metabolite immune correlates characteristic of immune responses following mucosal vaccination in calves. Male Holstein Friesian calves were vaccinated with Pfizer Rispoval® PI3 + RSV intranasal vaccine and metabolomic profiling of post-vaccination plasma revealed 12 metabolites whose peak intensities differed significantly from controls. Plasma levels of glycocholic acid, N-[(3α,5β,12α)-3,12-Dihydroxy-7,24-dioxocholan-24-yl]glycine, uric acid and