Dissertation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

CYP2A6) by P53
Transcriptional Regulation of Human Stress Responsive Cytochrome P450 2A6 (CYP2A6) by p53 Hao Hu M.Biotech. (Biotechnology) 2012 The University of Queensland B.B.A. 2009 University of Electronic Science and Technology of China B.Sc. (Pharmacy) 2009 University of Electronic Science and Technology of China A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2016 School of Medicine ABSTRACT Human cytochrome P450 (CYP) 2A6 is highly expressed in the liver and the encoding gene is regulated by various stress activated transcription factors, such as the nuclear factor (erythroid-derived 2)-like 2 (Nrf-2). Unlike the other xenobiotic metabolising CYP enzymes (XMEs), CYP2A6 only plays a minor role in xenobiotic metabolism. The CYP2A6 is highly induced by multiple forms of cellular stress conditions, where XMEs expression is normally inhibited. Recent findings suggest that the CYP2A6 plays an important role in regulating BR homeostasis. A computer based sequence analysis on the 3 kb proximate CYP2A6 promoter revealed several putative binding sites for p53, a protein that mediates regulation of antioxidant and apoptosis pathways. In this study, the role of p53 in CYP2A6 gene regulation is demonstrated. The site closest to transcription start site (TSS) is highly homologous with the p53 consensus sequence. The p53 responsiveness of this site was confirmed by transfections with various stepwise deleted of CYP2A6-5’-Luc constructs containing the putative p53RE. Deletion of the putative p53RE resulted in a total abolishment of p53 responsiveness of CYP2A6 promoter. Specific binding of p53 to the putative p53RE was detected by electrophoresis mobility shift assay. -

Characterisation of Bilirubin Metabolic Pathway in Hepatic Mitochondria Siti Nur Fadzilah Muhsain M.Sc
Characterisation of Bilirubin Metabolic Pathway in Hepatic Mitochondria Siti Nur Fadzilah Muhsain M.Sc. (Medical Research) 2005 Universiti Sains Malaysia Postgrad. Dip. (Toxicology) 2003 University of Surrey B.Sc.(Biomed. Sc.) 2000 Universiti Putra Malaysia A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2014 School of Medicine ABSTRACT Bilirubin (BR), a toxic waste product of degraded haem, is a potent antioxidant at physiological concentrations. To achieve the maximum benefit of BR, its intracellular level needs to be carefully regulated. A system comprising of two enzymes, haem oxygenase-1 (HMOX1) and cytochrome P450 2A5 (CYP2A5) exists in the endoplasmic reticulum (ER), responsible for regulating BR homeostasis. This system is induced in response to oxidative stress. In this thesis, oxidative stress caused accumulation of these enzymes in mitochondria — major producers and targets of reactive oxygen species (ROS) — is demonstrated. To understand the significance of this intracellular targeting, properties of microsomal and mitochondrial BR metabolising enzymes were compared and the capacity of mitochondrial CYP2A5 to oxidise BR in response to oxidative stress is reported. Microsomes and mitochondrial fractions were isolated from liver homogenates of DBA/2J mice, administered with sub-toxic dose of pyrazole, an oxidant stressor. The purity of extracted organelles was determined by analysing the expressions and activities of their respective marker enzymes. HMOX1 and CYP2A5 were significantly increased in microsomes and even more so in mitochondria in response to pyrazole-induced oxidative stress. By contrast, the treatment did not increase either microsomes or mitochondrial Uridine-diphosphate-glucuronosyltransferase 1A1 (UGT1A1), the sole enzyme that catalyses BR elimination through glucuronidation. -

Solarbio Catalogue with PRICES
CAS Name Grade Purity Biochemical Reagent Biochemical Reagent 75621-03-3 C8390-1 3-((3-Cholamidopropyl)dimethylammonium)-1-propanesulfonateCHAPS Ultra Pure Grade 1g 75621-03-3 C8390-5 3-((3-Cholamidopropyl)dimethylammonium)-1-propanesulfonateCHAPS 5g 57-09-0 C8440-25 Cetyl-trimethyl Ammonium Bromide CTAB High Pure Grade ≥99.0% 25g 57-09-0 C8440-100 Cetyl-trimethyl Ammonium Bromide CTAB High Pure Grade ≥99.0% 100g 57-09-0 C8440-500 Cetyl-trimethyl Ammonium Bromide CTAB High Pure Grade ≥99.0% 500g E1170-100 0.5M EDTA (PH8.0) 100ml E1170-500 0.5M EDTA (PH8.0) 500ml 6381-92-6 E8030-100 EDTA disodium salt dihydrate EDTA Na2 Biotechnology Grade ≥99.0% 100g 6381-92-6 E8030-500 EDTA disodium salt dihydrate EDTA Na2 Biotechnology Grade ≥99.0% 500g 6381-92-6 E8030-1000 EDTA disodium salt dihydrate EDTA Na2 Biotechnology Grade ≥99.0% 1kg 6381-92-6 E8030-5000 EDTA disodium salt dihydrate EDTA Na2 Biotechnology Grade ≥99.0% 5kg 60-00-4 E8040-100 Ethylenediaminetetraacetic acid EDTA Ultra Pure Grade ≥99.5% 100g 60-00-4 E8040-500 Ethylenediaminetetraacetic acid EDTA Ultra Pure Grade ≥99.5% 500g 60-00-4 E8040-1000 Ethylenediaminetetraacetic acid EDTA Ultra Pure Grade ≥99.5% 1kg 67-42-5 E8050-5 Ethylene glycol-bis(2-aminoethylether)-N,N,NEGTA′,N′-tetraacetic acid Ultra Pure Grade ≥97.0% 5g 67-42-5 E8050-10 Ethylene glycol-bis(2-aminoethylether)-N,N,NEGTA′,N′-tetraacetic acid Ultra Pure Grade ≥97.0% 10g 50-01-1 G8070-100 Guanidine Hydrochloride Guanidine HCl ≥98.0%(AT) 100g 50-01-1 G8070-500 Guanidine Hydrochloride Guanidine HCl ≥98.0%(AT) 500g 56-81-5 -

1 Molecular Physiology and Pathophysiology of Bilirubin Handling by the Blood, Liver
1 1 MOLECULAR PHYSIOLOGY AND PATHOPHYSIOLOGY OF BILIRUBIN HANDLING BY THE BLOOD, LIVER, 2 INTESTINE, AND BRAIN IN THE NEWBORN 3 THOR W.R. HANSEN1, RONALD J. WONG2, DAVID K. STEVENSON2 4 1Division of Paediatric and Adolescent Medicine, Institute of Clinical Medicine, Faculty of Medicine, 5 University of Oslo, Norway 6 2Department of Pediatrics, Stanford University School of Medicine, Stanford CA, USA 7 __________________________________________________________________________________ 8 2 9 I. Introduction 10 II. Bilirubin in the Body 11 A. Bilirubin Chemistry 12 1. Bilirubin structure 13 2. Bilirubin solubility 14 3. Bilirubin isomers 15 4. Heme degradation 16 5. Biliverdin and biliverdin reductase (BVR) 17 B. Bilirubin as an Antioxidant 18 C. Bilirubin as a Toxin 19 1. Bilirubin effects on enzyme activity 20 2. Toxicity of bilirubin conjugates and isomers 21 D. Other Functions/Roles 22 1. Drug displacement by bilirubin 23 2. Bilirubin interactions with the immune system and 24 inflammatory/infectious mechanisms 25 III. The Production of Bilirubin in the Body 26 A. Heme Catabolism and Its Regulation 27 1. Genetic variants in bilirubin production 28 B. The Effect of Hemolysis 29 1. Disorders associated with increased bilirubin production 30 IV. Bilirubin Binding and Transport in Blood 31 V. Bilirubin in the Liver 32 A. Hepatocellular Uptake and Intracellular Processing 33 B. Bilirubin Conjugation 34 1. Genetic variants in bilirubin conjugation 3 35 a. Crigler-Najjar syndrome type I 36 b. Crigler-Najjar syndrome type II 37 c. Gilbert syndrome 38 2. Genetic variants in transporter proteins 39 C. Bilirubin Excretion 40 VI. Bilirubin in the Intestines 41 A. -

Junbai Li Editor Supramolecular Chemistry of Biomimetic Systems Supramolecular Chemistry of Biomimetic Systems Junbai Li Editor
Junbai Li Editor Supramolecular Chemistry of Biomimetic Systems Supramolecular Chemistry of Biomimetic Systems Junbai Li Editor Supramolecular Chemistry of Biomimetic Systems 123 Editor Junbai Li Institute of Chemistry, Chinese Academy of Sciences Beijing China ISBN 978-981-10-6058-8 ISBN 978-981-10-6059-5 (eBook) DOI 10.1007/978-981-10-6059-5 Library of Congress Control Number: 2017949116 © Springer Nature Singapore Pte Ltd. 2017 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. The publisher, the authors and the editors are safe to assume that the advice and information in this book are believed to be true and accurate at the date of publication. Neither the publisher nor the authors or the editors give a warranty, express or implied, with respect to the material contained herein or for any errors or omissions that may have been made. The publisher remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Printed on acid-free paper This Springer imprint is published by Springer Nature The registered company is Springer Nature Singapore Pte Ltd. -

Generated by SRI International Pathway Tools Version 25.0 on Sat
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114625Cyc: Granulicella pectinivorans DSM 21001 Cellular Overview Connections between pathways are omitted for legibility. -
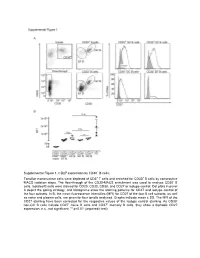
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

Resveratrol Is a Nuclear Receptor 4A1 (NR4A1) Ligand the Antagonizes NR4A1-Regulated Prooncogenic Pathways in Lung Cancers
Resveratrol is a Nuclear Receptor 4A1 (NR4A1) Ligand the Antagonizes NR4A1-Regulated Prooncogenic Pathways in Lung Cancers Lei Zhang1, Kumaravel Mohankumar1, Gregory Martin1, and Stephen Safe1 1Department of Veterinary Physiology and Pharmacology, Texas A&M University, College Station, TX, 77843 Abstract Methods A. C. Resveratrol (3,5,4’-trihydroxystilbene) is a polyphenolic phytochemical found in fruits, nuts, Ø Cell line: A549, H460 and H1299 non-small lung cancer cell lines were purchased and vegetables and there is evidence that this compound offers protection from several from American Type Culture Collection. human diseases including cancer. In cancer cell lines, resveratrol inhibits cell growth, survival, Ø Cell proliferation Assay: A549, H460, H1299 cells are seeded (2x104 per well) in migration/invasion and genes/pathways associated with these anticancer activities. Many of 96-well plates. A549 cells were maintained with DEME and 10% FBS. H460 and B. the same anticancer activities reported for resveratrol have previously been observed in this H1299 cells were maintained in RPMI 1640 and 10% FBS. Cells were allowed to laboratory using bis-indole derived nuclear receptor 4A1 (NR4A1) ligands that antagonize of attach for 24 hours. Then, the cells were treated at given concentrations with NR4A1-regulated pro-oncogenic pathways. Treatment of A549, H460, H1299 lung cancer cells appropriate media and 2.5% stripe FBS for 24, 48, and 72 hours. XTT cell viability with 50-125 µM resveratrol for 24, 48, 72 hours inhibited cell growth and IC50 values for kit that was purchased from Cell Signaling was applied to stain live cells after growth inhibition decreased with time. -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -

Figure S1. Gene Ontology Classification of Abeliophyllum Distichum Leaves Extract-Induced Degs
Figure S1. Gene ontology classification of Abeliophyllum distichum leaves extract-induced DEGs. The results are summarized in three main categories: Biological process, Cellular component and Molecular function. Figure S2. KEGG pathway enrichment analysis using Abeliophyllum distichum leaves extract-DEGs (A). Venn diagram analysis of DEGs involved in PI3K/Akt signaling pathway and Rap1 signaling pathway (B). Figure S3. The expression (A) and protein levels (B) of Akt3 in AL-treated SK-MEL2 cells. Values with different superscripted letters are significantly different (p < 0.05). Table S1. Abeliophyllum distichum leaves extract-induced DEGs. log2 Fold Gene name Gene description Change A2ML1 alpha-2-macroglobulin-like protein 1 isoform 2 [Homo sapiens] 3.45 A4GALT lactosylceramide 4-alpha-galactosyltransferase [Homo sapiens] −1.64 ABCB4 phosphatidylcholine translocator ABCB4 isoform A [Homo sapiens] −1.43 ABCB5 ATP-binding cassette sub-family B member 5 isoform 1 [Homo sapiens] −2.99 ABHD17C alpha/beta hydrolase domain-containing protein 17C [Homo sapiens] −1.62 ABLIM2 actin-binding LIM protein 2 isoform 1 [Homo sapiens] −2.53 ABTB2 ankyrin repeat and BTB/POZ domain-containing protein 2 [Homo sapiens] −1.48 ACACA acetyl-CoA carboxylase 1 isoform 1 [Homo sapiens] −1.76 ACACB acetyl-CoA carboxylase 2 precursor [Homo sapiens] −2.03 ACSM1 acyl-coenzyme A synthetase ACSM1, mitochondrial [Homo sapiens] −3.05 disintegrin and metalloproteinase domain-containing protein 19 preproprotein [Homo ADAM19 −1.65 sapiens] disintegrin and metalloproteinase -

From Heterogeneous and Extracellular Electron Transfer to Applications in Aqueous Batteries and Biofuel Cells
UNIVERSIDADE DE SÃO PAULO INSTITUTO DE QUÍMICA DE SÃO CARLOS PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA Graziela Cristina Sedenho Bioelectrochemical and bioinspired energy conversion systems: from heterogeneous and extracellular electron transfer to applications in aqueous batteries and biofuel cells São Carlos – SP 2021 GRAZIELA CRISTINA SEDENHO Bioelectrochemical and bioinspired energy conversion systems: from heterogeneous and extracellular electron transfer to applications in aqueous batteries and biofuel cells Thesis presented to the São Carlos Institute of Chemistry (IQSC), University of São Paulo, in partial fulfillment of the requirements for the degree of Doctor of Science. Concentration area: Physical Chemistry Advisor: Prof. Dr. Frank Nelson Crespilho Exemplar revisado O exemplar original encontra-se em acervo reservado na Biblioteca do IQSC-USP São Carlos – SP 2021 Autorizo a reprodução e divulgação total ou parcial deste trabalho, por qualquer meio convencional ou eletrônico para fins de estudo e pesquisa, desde que citada a fonte. Assinatura: Data: 06/01/2021 Ficha Catalográfica elaborada pela Seção de Referência e Atendimento ao Usuário do SBI/IQSC Sedenho, Graziela Cristina Bioelectrochemical and bioinspired energy conversion systems: from heterogeneous and extracellular electron transfer to applications in aqueous batteries and biofuel cells / Graziela Cristina Sedenho. — São Carlos, 2021. 137 f. Tese (Doutorado em Físico-Química) — Instituto de Química de São Carlos / Universidade de São Paulo, 2021. Orientador: Prof. Dr. Frank Nelson Crespilho 1. Transformação de energia. 2. Eletrodos de carbono. 3. Imobilização enzimática. 4. Reação de redução de oxigênio. 5. Transferência extracelular de elétrons. I. Título. Wilneide do Carmo Marchi Maiorano - CRB: 3978/8 ACKNOWLEDGMENTS • To my parents, Luís and Sueli, for teaching me to be strong, for always supporting and encouraging me.