Update on Pathogenesis of Primary
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae
Journal of Fungi Article Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae Mohammad Sayari 1,2,*, Magrieta A. van der Nest 1,3, Emma T. Steenkamp 1, Saleh Rahimlou 4 , Almuth Hammerbacher 1 and Brenda D. Wingfield 1 1 Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (A.H.); brenda.wingfi[email protected] (B.D.W.) 2 Department of Plant Science, University of Manitoba, 222 Agriculture Building, Winnipeg, MB R3T 2N2, Canada 3 Biotechnology Platform, Agricultural Research Council (ARC), Onderstepoort Campus, Pretoria 0110, South Africa 4 Department of Mycology and Microbiology, University of Tartu, 14A Ravila, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Fax: +1-204-474-7528 Abstract: Terpenes represent the biggest group of natural compounds on earth. This large class of organic hydrocarbons is distributed among all cellular organisms, including fungi. The different classes of terpenes produced by fungi are mono, sesqui, di- and triterpenes, although triterpene ergosterol is the main sterol identified in cell membranes of these organisms. The availability of genomic data from members in the Ceratocystidaceae enabled the detection and characterization of the genes encoding the enzymes in the mevalonate and ergosterol biosynthetic pathways. Using Citation: Sayari, M.; van der Nest, a bioinformatics approach, fungal orthologs of sterol biosynthesis genes in nine different species M.A.; Steenkamp, E.T.; Rahimlou, S.; of the Ceratocystidaceae were identified. -

The Impact of Transcription on Metabolism in Prostate and Breast Cancers
25 9 Endocrine-Related N Poulose et al. From hormones to fats and 25:9 R435–R452 Cancer back REVIEW The impact of transcription on metabolism in prostate and breast cancers Ninu Poulose1, Ian G Mills1,2,* and Rebecca E Steele1,* 1Centre for Cancer Research and Cell Biology, Queen’s University of Belfast, Belfast, UK 2Nuffield Department of Surgical Sciences, John Radcliffe Hospital, University of Oxford, Oxford, UK Correspondence should be addressed to I G Mills: [email protected] *(I G Mills and R E Steele contributed equally to this work) Abstract Metabolic dysregulation is regarded as an important driver in cancer development and Key Words progression. The impact of transcriptional changes on metabolism has been intensively f androgen studied in hormone-dependent cancers, and in particular, in prostate and breast cancer. f androgen receptor These cancers have strong similarities in the function of important transcriptional f breast drivers, such as the oestrogen and androgen receptors, at the level of dietary risk and f oestrogen epidemiology, genetics and therapeutically. In this review, we will focus on the function f endocrine therapy of these nuclear hormone receptors and their downstream impact on metabolism, with a resistance particular focus on lipid metabolism. We go on to discuss how lipid metabolism remains dysregulated as the cancers progress. We conclude by discussing the opportunities that this presents for drug repurposing, imaging and the development and testing of new Endocrine-Related Cancer therapeutics and treatment combinations. (2018) 25, R435–R452 Introduction: prostate and breast cancer Sex hormones act through nuclear hormone receptors Early-stage PCa is dependent on androgens for survival and induce distinct transcriptional programmes essential and can be treated by androgen deprivation therapy; to male and female physiology. -

33 34 35 Lipid Synthesis Laptop
BI/CH 422/622 Liver cytosol ANABOLISM OUTLINE: Photosynthesis Carbohydrate Biosynthesis in Animals Biosynthesis of Fatty Acids and Lipids Fatty Acids Triacylglycerides contrasts Membrane lipids location & transport Glycerophospholipids Synthesis Sphingolipids acetyl-CoA carboxylase Isoprene lipids: fatty acid synthase Ketone Bodies ACP priming 4 steps Cholesterol Control of fatty acid metabolism isoprene synth. ACC Joining Reciprocal control of b-ox Cholesterol Synth. Diversification of fatty acids Fates Eicosanoids Cholesterol esters Bile acids Prostaglandins,Thromboxanes, Steroid Hormones and Leukotrienes Metabolism & transport Control ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Synthesis Degradation Ketone body Utilization Isoprene Biosynthesis 1 Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates are transferred stepwise from ATP to mevalonate. • A third phosphate from ATP is added at the hydroxyl, followed by decarboxylation and elimination catalyzed by pyrophospho- mevalonate decarboxylase creates a pyrophosphorylated 5-C product: D3-isopentyl pyrophosphate (IPP) (isoprene). • Isomerization to a second isoprene dimethylallylpyrophosphate (DMAPP) gives two activated isoprene IPP compounds that act as precursors for D3-isopentyl pyrophosphate Isopentyl-D-pyrophosphate all of the other lipids in this class isomerase DMAPP Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates -

A Key Mammalian Cholesterol Synthesis Enzyme, Squalene Monooxygenase, Is Allosterically Stabilized by Its Substrate
A key mammalian cholesterol synthesis enzyme, squalene monooxygenase, is allosterically stabilized by its substrate Hiromasa Yoshiokaa, Hudson W. Coatesb, Ngee Kiat Chuab, Yuichi Hashimotoa, Andrew J. Brownb,1, and Kenji Ohganea,c,1 aInstitute for Quantitative Biosciences, The University of Tokyo, 113-0032 Tokyo, Japan; bSchool of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW 2052, Australia; and cSynthetic Organic Chemistry Laboratory, RIKEN, 351-0198 Saitama, Japan Edited by Brenda A. Schulman, Max Planck Institute of Biochemistry, Martinsried, Germany, and approved February 18, 2020 (received for review September 20, 2019) Cholesterol biosynthesis is a high-cost process and, therefore, is the cognate E3 ligase for SM (9, 10), catalyzing its ubiquitination tightly regulated by both transcriptional and posttranslational in the N100 region (11). negative feedback mechanisms in response to the level of cellular Despite its importance in cholesterol-accelerated degradation, cholesterol. Squalene monooxygenase (SM, also known as squalene the structure and function of the SM-N100 region has not been epoxidase or SQLE) is a rate-limiting enzyme in the cholesterol fully resolved. Thus far, biochemical analyses have revealed the biosynthetic pathway and catalyzes epoxidation of squalene. The presence of a reentrant loop anchoring SM to the ER membrane stability of SM is negatively regulated by cholesterol via its and an amphipathic helix required for cholesterol-accelerated N-terminal regulatory domain (SM-N100). In this study, using a degradation (7, 8), while X-ray crystallography has revealed the SM-luciferase fusion reporter cell line, we performed a chemical architecture of the catalytic domain of SM (12). However, the genetics screen that identified inhibitors of SM itself as up-regulators highly hydrophobic and disordered nature of the N100 region of SM. -

Integrating Data Mining, Network Pharmacology, and Molecular
Integrating Data Mining, Network Pharmacology, and Molecular Docking Verication to Investigate the Molecular Mechanism of Traditional Chinese Medicine Prescriptions for Treating Male Infertility Xue Bai Beijing University of Chinese Medicine Yibo Tang Beijing University of Chinese Medicine Qiang Li Beijing University of Chinese Medicine Guimin Liu Beijing University of Chinese Medicine Dan Liu Beijing University of Chinese Medicine Xiaolei Fan Beijing University of Chinese Medicine Zhejun Liu Beijing University of Chinese Medicine Shujun Yu Beijing University of Chinese Medicine Tian Tang Beijing University of Chinese Medicine Shuyan Wang Beijing University of Chinese Medicine Lingru Li Beijing University of Chinese Medicine Kailin Zhou Beijing University of Chinese Medicine Yanfei Zheng Beijing University of Chinese Medicine Zhenquan Liu ( [email protected] ) Research Page 1/42 Keywords: Traditional Chinese medicine, male infertility, data mining, network pharmacology, molecular docking Posted Date: March 4th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-264555/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/42 Abstract Background: Male infertility (MI) affects almost 5% adult men worldwide, and 75% of these cases are unexplained idiopathic. There are limitations in the current treatment due to the unclear mechanism of MI, which highlight the urgent need for a more effective strategy or drug. Traditional Chinese Medicine (TCM) prescriptions have been used to treat MI for thousands of years, but their molecular mechanism is not well dened. Methods: Aiming at revealing the molecular mechanism of TCM prescriptions on MI, a comprehensive strategy integrating data mining, network pharmacology, and molecular docking verication was performed. -
Figures Intermediary Ontogeny.Pptx
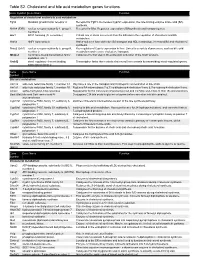
Table S2. Cholesterol and bile acid metabolism genes functions. Gene Symbol Gene Name Function Regulation of cholesterol and/or bile acid metabolism Fgfr4 fibroblast growth factor receptor 4 Receptor for Fgf15. Decreases Cyp7a1 expression, the rate-limiting enzyme in bile acid (BA) synthesis. Nr1h4 (FXR) nuclear receptor subfamily 1, group H, Receptor for BAs. Regulates expression of BAsynthesis and transport genes. member 4 Arv1 ARV1 homolog (S. cerevisiae) Critical role in sterol movement from the ER and in the regulation of cholesterol and BA metabolism. Hnf1a HNF1 homeobox A Hnf1a-null mice have defective BA transport and HDL metabolism, increased BA and cholesterol synthesis. Nr5a2 (Lrh1) nuclear receptor subfamily 5, group A, Key regulator of Cyp7a expression in liver. Linked to a variety of processes, such as bile acid member 2 metabolism and reverse cholesterol transport. Mbtps1 membrane-bound transcription factor Catalyzes the first step in the proteolytic activation of the Srebf proteins. peptidase, site 1 Srebf2 sterol regulatory element binding Transcription factor that controls cholesterol homeostasis by transcribing sterol-regulated genes. transcription factor 2 Gene Gene Name Function Symbol Bile acid metabolism Akr1c6 aldo-keto reductase family 1, member C1 May have a role in the transport and intrahepatic concentration of bile acids. Akr1d1 aldo-keto reductase family 1, member D1 Reduces BA intermediates 7-α,12-α-dihydroxy-4-cholesten-3-one & 7-α-hydroxy-4-cholesten-3-one. Amacr alpha-methylacyl-CoA racemase Responsible for the conversion of pristanoyl-CoA and C27-bile acyl-CoAs to their (S)-stereoisomers. Baat (Bat) bile acid CoA: amino acid N- Conjugates C24 bile acids to glycine or taurine before excretion into bile canaliculi. -

Open Full Page
Published OnlineFirst May 7, 2019; DOI: 10.1158/0008-5472.CAN-18-2529 Cancer Molecular Cell Biology Research Upregulation of Scavenger Receptor B1 Is Required for Steroidogenic and Nonsteroidogenic Cholesterol Metabolism in Prostate Cancer Jacob A. Gordon1,2, Jake W. Noble1, Ankur Midha3, Fatemeh Derakhshan4, Gang Wang4, Hans H. Adomat1, Emma S. Tomlinson Guns1,2, Yen-Yi Lin1, Shancheng Ren5, Collin C. Collins1, Peter S. Nelson6, Colm Morrissey7, Kishor M. Wasan2,8, and Michael E. Cox1,2,9 Abstract Aberrant cholesterol metabolism is increasingly appre- viability and induced endoplasmic reticulum stress and ciated to be essential for prostate cancer initiation and autophagy. The inability of exogenous steroids to reverse progression. Transcript expression of the high-density lipo- these effects indicates that AR pathway activation is insuffi- protein-cholesterol receptor scavenger receptor B1 (SR-B1) cient to overcome cytotoxic stress caused by a decrease in the is elevated in primary prostate cancer. Hypothesizing that availability of cholesterol. Furthermore, SR-B1 antagonism SR-B1 expression may help facilitate malignant transforma- decreased cholesterol uptake, growth, and viability of the tion, we document increased SR-B1 protein and transcript AR-null CRPC cell model PC-3, and the small-molecule SR- expression in prostate cancer relative to normal prostate B1 antagonist block lipid transport-1 decreased xenograft epithelium that persists in lethal castration-resistant prostate growth rate despite poor pharmacologic properties. Overall, cancer (CRPC) metastasis. As intratumoral steroid synthesis our findings show that SR-B1 is upregulated in primary and from the precursor cholesterol can drive androgen receptor castration-resistant disease and is essential for cholesterol (AR) pathway activity in CRPC, we screened androgenic uptake needed to drive both steroidogenic and nonsteroi- benign and cancer cell lines for sensitivity to SR-B1 antag- dogenic biogenic pathways, thus implicating SR-B1 as a onism. -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc. -

Trigonella Foenum-Graecum L.)
Journal of Medicinal Plants Bioengineering of Important Secondary Metabolites and Metabolic Pathways in Fenugreek (Trigonella foenum-graecum L.) 1* 2 3 Mehrafarin A (Ph.D. Student)1 , Qaderi A (Ph.D. Student) , 4Rezazadeh Sh (Ph.D.)5 , Naghdi Badi H (Ph.D.) , Noormohammadi Gh (Ph.D.) , Zand E (Ph.D.) 1- Department of Cultivation and Development, Institute of Medicinal Plants, ACECR, Karaj, Iran 2- Department of Plant Biotechnology, Institute of Medicinal Plants, ACECR, Karaj, Iran 3- Department of Pharmacognosy and Pharmaceutics, Institute of Medicinal Plants, ACECR, Karaj, Iran 4- Department of Agronomy, Science and Research Branch, Islamic Azad University (IAU), Tehran, Iran 5- Department of Weed Research, Iranian Plant Protection Research Institute, Tehran, Iran *Corresponding author: Culture and Research (ACECR), Kavosh Boulevard, Supa Boulevard, 55th Kilometer of Tehran-Gazvin Freeway, Pouleh Kordan, Karaj, Iran Tel: +98-261-4764010-19, Fax: +98-261- 4764021 Email: [email protected] Receive: 21 Jul. 2010 Acceptance: 4 Sep. 2010 Abstract Fenugreek (Trigonella foenum-graecum L.) has a long and respected history of medicinal uses in Middle East and Persian medicine. The hypocholesterolaemic and hypoglycaemic effects of fenugreek were attributed to its major steroidal sapogenin, diosgenin and its major alkaloid, trigonelline. The knowledge of diosgenin and trigonelline biosynthesis is derived from studies of cholesterol and nicotinic acid production through acetyl-CoA→ mevalonate→ isopentenyl pyrophosphate→ squalene→ lanosterol→ cholesterol→ diosgenin and quinolinic acid→ nicotinamide adenine dinucleotide→ nicotinamide→ nicotinic acid→ trigonelline pathways, respectively. This paper reviews the secondary metabolites and metabolic pathways of diosgenin and trigonelline production in fenugreek as a medicinal plant and economical crop. Keywords:Archive Fenugreek, Secondary metabolites, of Metabolic SID Pathways, Diosgenin, Trigonelline Volume 9, No. -

Hepatic Cytochrome P450 Reductase-Null Mice As an Animal Model to Study Electron Transfer Pathways in Cholesterol Synthesis and Cyp2e1-Mediated Drug Metabolism
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2006 HEPATIC CYTOCHROME P450 REDUCTASE-NULL MICE AS AN ANIMAL MODEL TO STUDY ELECTRON TRANSFER PATHWAYS IN CHOLESTEROL SYNTHESIS AND CYP2E1-MEDIATED DRUG METABOLISM Li Li University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Li, Li, "HEPATIC CYTOCHROME P450 REDUCTASE-NULL MICE AS AN ANIMAL MODEL TO STUDY ELECTRON TRANSFER PATHWAYS IN CHOLESTEROL SYNTHESIS AND CYP2E1-MEDIATED DRUG METABOLISM" (2006). University of Kentucky Doctoral Dissertations. 417. https://uknowledge.uky.edu/gradschool_diss/417 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Li Li The Graduate School University of Kentucky 2006 HEPATIC CYTOCHROME P450 REDUCTASE-NULL MICE AS AN ANIMAL MODEL TO STUDY ELECTRON TRANSFER PATHWAYS IN CHOLESTEROL SYNTHESIS AND CYP2E1-MEDIATED DRUG METABOLISM ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements of the degree of Doctor of Philosophy in The Graduate School at the University of Kentucky By Li Li Lexington, Kentucky Director: Dr. Todd D. Porter, Associate Professor of Pharmaceutical Sciences College of Pharmacy 2006 Copyright © Li Li 2006 ABSTRACT OF DISSERTATION HEPATIC CYTOCHROME P450 REDUCTASE-NULL MICE AS AN ANIMAL MODEL TO STUDY ELECTRON TRANSFER PATHWAYS IN CHOLESTEROL SYNTHESIS AND CYP2E1-MEDIATED DRUG METABOLISM NADPH-cytochrome P450 reductase (CPR) is a flavoprotein containing both FAD and FMN and functions as the electron donor protein for several oxygenase enzymes found on the endoplasmic reticulum of eukaryotic cells, including cytochrome P450s involved in drug metabolism and cholesterol biosynthesis. -

WO 2017/011215 Al 19 January 2017 (19.01.2017) P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/011215 Al 19 January 2017 (19.01.2017) P O PCT (51) International Patent Classification: TX 75209 (US). THEODOROPOULOS, Pano; 8567 A61K 31/427 (2006.01) A61K 31/454 (2006.01) Forrest Hills Blvd., Dallas, TX 75218 (US). A61K 31/444 (2006.01) (74) Agent: LUBIT, Beverly, W.; Chiesa Shahinian & Gianto- (21) International Application Number: masi PC, One Boland Drive, West Orange, NJ 07052 (US). PCT/US20 16/04093 1 (81) Designated States (unless otherwise indicated, for every (22) International Filing Date: kind of national protection available): AE, AG, AL, AM, 5 July 20 16 (05.07.2016) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (25) Filing Language: English DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (26) Publication Language: English HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (30) Priority Data: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 62/193,019 15 July 2015 (15.07.2015) US PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (71) Applicant: THE BOARD OF REGENTS OF THE UNI¬ SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, VERSITY OF TEXAS SYSTEM [US/US]; 201 West 7th TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Alpha-Terpineol Production from an Engineered Saccharomyces
Zhang et al. Microb Cell Fact (2019) 18:160 https://doi.org/10.1186/s12934-019-1211-0 Microbial Cell Factories RESEARCH Open Access Alpha-Terpineol production from an engineered Saccharomyces cerevisiae cell factory Chuanbo Zhang1, Man Li1, Guang‑Rong Zhao1,2,3 and Wenyu Lu1,2,3* Abstract Background: Alpha‑Terpineol (α‑Terpineol), a C10 monoterpenoid alcohol, is widely used in the cosmetic and pharmaceutical industries. Construction Saccharomyces cerevisiae cell factories for producing monoterpenes ofers a promising means to substitute chemical synthesis or phytoextraction. Results: α‑Terpineol was produced by expressing the truncated α‑Terpineol synthase (tVvTS) from Vitis vinifera in S. cerevisiae. The α‑Terpineol titer was increased to 0.83 mg/L with overexpression of the rate‑limiting genes tHMG1, IDI1 and ERG20F96W-N127W. A GSGSGSGSGS linker was applied to fuse ERG20F96W‑N127W with tVvTS, and expressing the fusion protein increased the α‑Terpineol production by 2.87‑fold to 2.39 mg/L when compared with the parental strain. In addition, we found that farnesyl diphosphate (FPP) accumulation by down‑regulation of ERG9 expression and deletion of LPP1 and DPP1 did not improve α‑Terpineol production. Therefore, ERG9 was overexpressed and the α‑Terpineol titer was further increased to 3.32 mg/L. The best α‑Terpineol producing strain LCB08 was then used for batch and fed‑batch fermentation in a 5 L bioreactor, and the production of α‑Terpineol was ultimately improved to 21.88 mg/L. Conclusions: An efcient α‑Terpineol production cell factory was constructed by engineering the S. cerevisiae meva‑ lonate pathway, and the metabolic engineering strategies could also be applied to produce other valuable monoter‑ pene compounds in yeast.