Cholesterol-Dependent Degradation and Unsaturated Fatty Acid-Dependent Stabilisation of Squalene Monooxygenase in the Control of Cholesterol Synthesis
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae
Journal of Fungi Article Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae Mohammad Sayari 1,2,*, Magrieta A. van der Nest 1,3, Emma T. Steenkamp 1, Saleh Rahimlou 4 , Almuth Hammerbacher 1 and Brenda D. Wingfield 1 1 Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (A.H.); brenda.wingfi[email protected] (B.D.W.) 2 Department of Plant Science, University of Manitoba, 222 Agriculture Building, Winnipeg, MB R3T 2N2, Canada 3 Biotechnology Platform, Agricultural Research Council (ARC), Onderstepoort Campus, Pretoria 0110, South Africa 4 Department of Mycology and Microbiology, University of Tartu, 14A Ravila, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Fax: +1-204-474-7528 Abstract: Terpenes represent the biggest group of natural compounds on earth. This large class of organic hydrocarbons is distributed among all cellular organisms, including fungi. The different classes of terpenes produced by fungi are mono, sesqui, di- and triterpenes, although triterpene ergosterol is the main sterol identified in cell membranes of these organisms. The availability of genomic data from members in the Ceratocystidaceae enabled the detection and characterization of the genes encoding the enzymes in the mevalonate and ergosterol biosynthetic pathways. Using Citation: Sayari, M.; van der Nest, a bioinformatics approach, fungal orthologs of sterol biosynthesis genes in nine different species M.A.; Steenkamp, E.T.; Rahimlou, S.; of the Ceratocystidaceae were identified. -

The Impact of Transcription on Metabolism in Prostate and Breast Cancers
25 9 Endocrine-Related N Poulose et al. From hormones to fats and 25:9 R435–R452 Cancer back REVIEW The impact of transcription on metabolism in prostate and breast cancers Ninu Poulose1, Ian G Mills1,2,* and Rebecca E Steele1,* 1Centre for Cancer Research and Cell Biology, Queen’s University of Belfast, Belfast, UK 2Nuffield Department of Surgical Sciences, John Radcliffe Hospital, University of Oxford, Oxford, UK Correspondence should be addressed to I G Mills: [email protected] *(I G Mills and R E Steele contributed equally to this work) Abstract Metabolic dysregulation is regarded as an important driver in cancer development and Key Words progression. The impact of transcriptional changes on metabolism has been intensively f androgen studied in hormone-dependent cancers, and in particular, in prostate and breast cancer. f androgen receptor These cancers have strong similarities in the function of important transcriptional f breast drivers, such as the oestrogen and androgen receptors, at the level of dietary risk and f oestrogen epidemiology, genetics and therapeutically. In this review, we will focus on the function f endocrine therapy of these nuclear hormone receptors and their downstream impact on metabolism, with a resistance particular focus on lipid metabolism. We go on to discuss how lipid metabolism remains dysregulated as the cancers progress. We conclude by discussing the opportunities that this presents for drug repurposing, imaging and the development and testing of new Endocrine-Related Cancer therapeutics and treatment combinations. (2018) 25, R435–R452 Introduction: prostate and breast cancer Sex hormones act through nuclear hormone receptors Early-stage PCa is dependent on androgens for survival and induce distinct transcriptional programmes essential and can be treated by androgen deprivation therapy; to male and female physiology. -

33 34 35 Lipid Synthesis Laptop
BI/CH 422/622 Liver cytosol ANABOLISM OUTLINE: Photosynthesis Carbohydrate Biosynthesis in Animals Biosynthesis of Fatty Acids and Lipids Fatty Acids Triacylglycerides contrasts Membrane lipids location & transport Glycerophospholipids Synthesis Sphingolipids acetyl-CoA carboxylase Isoprene lipids: fatty acid synthase Ketone Bodies ACP priming 4 steps Cholesterol Control of fatty acid metabolism isoprene synth. ACC Joining Reciprocal control of b-ox Cholesterol Synth. Diversification of fatty acids Fates Eicosanoids Cholesterol esters Bile acids Prostaglandins,Thromboxanes, Steroid Hormones and Leukotrienes Metabolism & transport Control ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Synthesis Degradation Ketone body Utilization Isoprene Biosynthesis 1 Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates are transferred stepwise from ATP to mevalonate. • A third phosphate from ATP is added at the hydroxyl, followed by decarboxylation and elimination catalyzed by pyrophospho- mevalonate decarboxylase creates a pyrophosphorylated 5-C product: D3-isopentyl pyrophosphate (IPP) (isoprene). • Isomerization to a second isoprene dimethylallylpyrophosphate (DMAPP) gives two activated isoprene IPP compounds that act as precursors for D3-isopentyl pyrophosphate Isopentyl-D-pyrophosphate all of the other lipids in this class isomerase DMAPP Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates -

WO 2009/005519 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date PCT (10) International Publication Number 8 January 2009 (08.01.2009) WO 2009/005519 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 31/19 (2006.01) A61K 31/185 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 31/20 (2006.01) A61K 31/225 (2006.01) AT,AU, AZ, BA, BB, BG, BH, BR, BW, BY,BZ, CA, CH, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, (21) International Application Number: ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, PCT/US2007/072499 IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY,MA, MD, ME, MG, MK, MN, MW, (22) International Filing Date: 29 June 2007 (29.06.2007) MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, PT, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, SV, SY, (25) Filing Language: English TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW (26) Publication Language: English (71) Applicant (for all designated States except US): AC- (84) Designated States (unless otherwise indicated, for every CERA, INC. [US/US]; 380 Interlocken Crescent, Suite kind of regional protection available): ARIPO (BW, GH, 780, Broomfield, CO 80021 (US). GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), (72) Inventor; and European (AT,BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, (75) Inventor/Applicant (for US only): HENDERSON, FR, GB, GR, HU, IE, IS, IT, LT,LU, LV,MC, MT, NL, PL, Samuel, T. -

Squalene, Olive Oil, and Cancer Risk: a Review and Hypothesis
Vol. 6, 1101-1103, December 1997 Cancer Epidemiology,Blornarkers & Prevention 1101 Review Squalene, Olive Oil, and Cancer Risk: A Review and Hypothesis Harold L. Newmark’ of unsaturated fatty acids. In Italy, about 80% of edible oil is Strang Cancer Research Laboratory, The Rockefeller University, New York, olive oil,2 suggesting a protective effect of olive oil intake. In New York 10021, and Laboratory for Cancer Research, College of Pharmacy, another Italian case-control study of diet and pancreatic cancer Rutgers University, Piscataway, New Jersey 08855-0789 (8), increased frequency of edible oil consumption was associ- ated with a trend toward decreased risk. Edible oil in Italy consists of about 80% olive oil, thus suggesting a protective Abstract effect of olive oil in pancreatic cancer.2 Epidemioiogical studies of breast and pancreatic cancer Animal studies on fat and cancer have generally shown in several Mediterranean populations have demonstrated that olive oil either has no effect or a protective effect on the that increased dietary intake of olive oil is associated with prevention of a variety of chemically induced tumors. Olive oil a small decreased risk or no increased risk of cancer, did not increase tumor incidence or growth, in contrast to corn despite a higjier proportion of overall lipid intake. and sunflower oil, in some mammary cancer models (9 -1 1) and Experimental animal model studies of high dietary fat also in colon cancer models (12, 13), although in an earlier and cancer also indicate that olive oil has either no effect report, olive oil exhibited mammary tumor-promoting proper- or a protective effect on the prevention of a variety of ties similar to that ofcorn oil (14), in contrast to the later studies chemically induced tumors. -

Update on Pathogenesis of Primary
177:3 C E Flück Update primary 177:3 R99–R111 Review adrenal insufficiency MECHANISMS IN ENDOCRINOLOGY Update on pathogenesis of primary adrenal insufficiency: beyond steroid enzyme deficiency and autoimmune adrenal destruction Correspondence Christa E Flück should be addressed Departments of Pediatrics and Clinical Research, Bern University Children’s Hospital Inselspital, University of Bern, to C E Flück Bern, Switzerland Email [email protected] Abstract Primary adrenal insufficiency (PAI) is potentially life threatening, but rare. In children, genetic defects prevail whereas adults suffer more often from acquired forms of PAI. The spectrum of genetic defects has increased in recent years with the use of next-generation sequencing methods and now has reached far beyond genetic defects in all known enzymes of adrenal steroidogenesis. Cofactor disorders such as P450 oxidoreductase (POR) deficiency manifesting as a complex form of congenital adrenal hyperplasia with a broad clinical phenotype have come to the fore. In patients with isolated familial glucocorticoid deficiency (FGD), in which no mutations in the genes for the ACTH receptor (MC2R) or its accessory protein MRAP have been found, non-classic steroidogenic acute regulatory protein (StAR) and CYP11A1 mutations have been described; and more recently novel mutations in genes such as nicotinamide nucleotide transhydrogenase (NNT) and thioredoxin reductase 2 (TRXR2) involved in the maintenance of the mitochondrial redox potential and generation of NADPH important for steroidogenesis and ROS detoxication have been discovered. European Journal European of Endocrinology In addition, whole exome sequencing approach also solved the genetics of some syndromic forms of PAI including IMAGe syndrome (CDKN1C), Irish traveler syndrome (MCM4), MIRAGE syndrome (SAMD9); and most recently a syndrome combining FGD with steroid-resistant nephrotic syndrome and ichthyosis caused by mutations in the gene for sphingosine-1-phosphate lyase 1 (SGPL1). -

A Key Mammalian Cholesterol Synthesis Enzyme, Squalene Monooxygenase, Is Allosterically Stabilized by Its Substrate
A key mammalian cholesterol synthesis enzyme, squalene monooxygenase, is allosterically stabilized by its substrate Hiromasa Yoshiokaa, Hudson W. Coatesb, Ngee Kiat Chuab, Yuichi Hashimotoa, Andrew J. Brownb,1, and Kenji Ohganea,c,1 aInstitute for Quantitative Biosciences, The University of Tokyo, 113-0032 Tokyo, Japan; bSchool of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW 2052, Australia; and cSynthetic Organic Chemistry Laboratory, RIKEN, 351-0198 Saitama, Japan Edited by Brenda A. Schulman, Max Planck Institute of Biochemistry, Martinsried, Germany, and approved February 18, 2020 (received for review September 20, 2019) Cholesterol biosynthesis is a high-cost process and, therefore, is the cognate E3 ligase for SM (9, 10), catalyzing its ubiquitination tightly regulated by both transcriptional and posttranslational in the N100 region (11). negative feedback mechanisms in response to the level of cellular Despite its importance in cholesterol-accelerated degradation, cholesterol. Squalene monooxygenase (SM, also known as squalene the structure and function of the SM-N100 region has not been epoxidase or SQLE) is a rate-limiting enzyme in the cholesterol fully resolved. Thus far, biochemical analyses have revealed the biosynthetic pathway and catalyzes epoxidation of squalene. The presence of a reentrant loop anchoring SM to the ER membrane stability of SM is negatively regulated by cholesterol via its and an amphipathic helix required for cholesterol-accelerated N-terminal regulatory domain (SM-N100). In this study, using a degradation (7, 8), while X-ray crystallography has revealed the SM-luciferase fusion reporter cell line, we performed a chemical architecture of the catalytic domain of SM (12). However, the genetics screen that identified inhibitors of SM itself as up-regulators highly hydrophobic and disordered nature of the N100 region of SM. -

Inhibition of Sterol Biosynthesis by Bacitracin (Mevalonic Acid/CQ-Isopentenyl Pyrophosphate/C15-Farnesyl Pyrophosphate) K
Proc. Nat. Acad. Sci. USA Vol. 69, No. 5, pp. 1287-1289, May 1972 Inhibition of Sterol Biosynthesis by Bacitracin (mevalonic acid/CQ-isopentenyl pyrophosphate/C15-farnesyl pyrophosphate) K. JOHN STONE* AND JACK L. STROMINGERt Biological Laboratories, Harvard University, Cambridge, Massachusetts 02138 Contributed by Jack L. Strominger, March 14 1972 ABSTRACT Bacitracin is an inhibitor of the biosyn- muslin eliminated floating particles of fat from the enzyme thesis of squalene and sterols from mevalonic acid, C5- solution enzyme). In some the isopentenyl pyrophosphate, or C15-farnesyl pyrophosphate (Sio experiments, S10 enzyme catalyzed by preparations from rat liver. The antibiotic was centrifuged at 105,000 X g for 90 min to remove micro- is active at extremely low ratios of antibiotic to substrate. somal particles, and this supernatant is referred to as the Sc1t The mechanism of inhibition appears to be the formation enzyme. of a complex between bacitracin, divalent cation, and C15- Bacitracin was generously provided by the Upjohn Co., farnesyl pyrophosphate and other isoprenyl pyrophos- phates. It is similar to the formation of the complex with Kalamazoo, Mich. [2-14C]3RS-mevalonic acid was obtained C55-isopropenyl pyrophosphate in microbial systems. The from New England Nuclear Co., [SH]farnesyl pyrophosphate toxicity of bacitracin for animal cells could be due in part and [2-'4C]isopentenyl pyrophosphate were generous gifts to the formation of these complexes. from Dr. Konrad Bloch. The Enzyme incubations (about 0.5-ml samples) were termi- antibacterial properties of bacitracin result, in part, from nated by in a water bath for 1 an inhibition of the heating boiling min. -

Livers of Sharks, Especially Deepwater Shark Species Which Are Especially Vulnerable Theto Overfishing and Often Endangered
AN EXCLUSIVE STUDY BY BLOOM March 2015 Shark in our beauty BEAST creams and the Beauty Shark in our beauty BEAST creams Some cosmetic brands still use a substance derived from the liver of sharks in the blends of moisturizing creams. In 2012,and BLOOM carried out a worldwide study on the use of shark squalane, a substance extracted from the livers of sharks, especially deepwater shark species which are especially vulnerable theto overfishing and often endangered. The study revealed that the cosmetics sector was the main consumer of animal squalane, even though plant substitutes derived from sugar cane or olive were available. The report pointed to the urgency for corporations to take their environmental responsibility seriously and to modify their supply chain in order to exclusively use plant squalane. Two years later, in 2014, BLOOM proceeded to carry out the largest test ever made on commercial creams to identify the presence of shark squalane in them. In total, BLOOM tested 72 moisturizing creams whose list of ingredients mentioned “squalane”. Labels did not specify whether the squalane was animal-based (shark) or plant-based (olive or sugarcane). Ten creams out of 72 did not contain a sufficient volume of squalane altogether to identify its origin, but results were robust for 62 of the products tested. As a conclusion, one moisturizing cream out of five contains shark squalane. It appearsBeauty that a high proportion of Asian brands still use shark squalane while Western brands, although still massively concerned by this issue in 2012, have globally shifted away from using shark substances. -

Steroidal Triterpenes of Cholesterol Synthesis
Molecules 2013, 18, 4002-4017; doi:10.3390/molecules18044002 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Steroidal Triterpenes of Cholesterol Synthesis Jure Ačimovič and Damjana Rozman * Centre for Functional Genomics and Bio-Chips, Faculty of Medicine, Institute of Biochemistry, University of Ljubljana, Zaloška 4, Ljubljana SI-1000, Slovenia; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +386-1-543-7591; Fax: +386-1-543-7588. Received: 18 February 2013; in revised form: 19 March 2013 / Accepted: 27 March 2013 / Published: 4 April 2013 Abstract: Cholesterol synthesis is a ubiquitous and housekeeping metabolic pathway that leads to cholesterol, an essential structural component of mammalian cell membranes, required for proper membrane permeability and fluidity. The last part of the pathway involves steroidal triterpenes with cholestane ring structures. It starts by conversion of acyclic squalene into lanosterol, the first sterol intermediate of the pathway, followed by production of 20 structurally very similar steroidal triterpene molecules in over 11 complex enzyme reactions. Due to the structural similarities of sterol intermediates and the broad substrate specificity of the enzymes involved (especially sterol-Δ24-reductase; DHCR24) the exact sequence of the reactions between lanosterol and cholesterol remains undefined. This article reviews all hitherto known structures of post-squalene steroidal triterpenes of cholesterol synthesis, their biological roles and the enzymes responsible for their synthesis. Furthermore, it summarises kinetic parameters of enzymes (Vmax and Km) and sterol intermediate concentrations from various tissues. Due to the complexity of the post-squalene cholesterol synthesis pathway, future studies will require a comprehensive meta-analysis of the pathway to elucidate the exact reaction sequence in different tissues, physiological or disease conditions. -

Integrating Data Mining, Network Pharmacology, and Molecular
Integrating Data Mining, Network Pharmacology, and Molecular Docking Verication to Investigate the Molecular Mechanism of Traditional Chinese Medicine Prescriptions for Treating Male Infertility Xue Bai Beijing University of Chinese Medicine Yibo Tang Beijing University of Chinese Medicine Qiang Li Beijing University of Chinese Medicine Guimin Liu Beijing University of Chinese Medicine Dan Liu Beijing University of Chinese Medicine Xiaolei Fan Beijing University of Chinese Medicine Zhejun Liu Beijing University of Chinese Medicine Shujun Yu Beijing University of Chinese Medicine Tian Tang Beijing University of Chinese Medicine Shuyan Wang Beijing University of Chinese Medicine Lingru Li Beijing University of Chinese Medicine Kailin Zhou Beijing University of Chinese Medicine Yanfei Zheng Beijing University of Chinese Medicine Zhenquan Liu ( [email protected] ) Research Page 1/42 Keywords: Traditional Chinese medicine, male infertility, data mining, network pharmacology, molecular docking Posted Date: March 4th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-264555/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/42 Abstract Background: Male infertility (MI) affects almost 5% adult men worldwide, and 75% of these cases are unexplained idiopathic. There are limitations in the current treatment due to the unclear mechanism of MI, which highlight the urgent need for a more effective strategy or drug. Traditional Chinese Medicine (TCM) prescriptions have been used to treat MI for thousands of years, but their molecular mechanism is not well dened. Methods: Aiming at revealing the molecular mechanism of TCM prescriptions on MI, a comprehensive strategy integrating data mining, network pharmacology, and molecular docking verication was performed. -
Figures Intermediary Ontogeny.Pptx
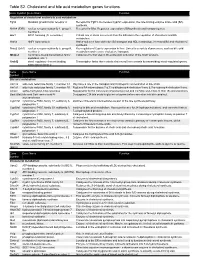
Table S2. Cholesterol and bile acid metabolism genes functions. Gene Symbol Gene Name Function Regulation of cholesterol and/or bile acid metabolism Fgfr4 fibroblast growth factor receptor 4 Receptor for Fgf15. Decreases Cyp7a1 expression, the rate-limiting enzyme in bile acid (BA) synthesis. Nr1h4 (FXR) nuclear receptor subfamily 1, group H, Receptor for BAs. Regulates expression of BAsynthesis and transport genes. member 4 Arv1 ARV1 homolog (S. cerevisiae) Critical role in sterol movement from the ER and in the regulation of cholesterol and BA metabolism. Hnf1a HNF1 homeobox A Hnf1a-null mice have defective BA transport and HDL metabolism, increased BA and cholesterol synthesis. Nr5a2 (Lrh1) nuclear receptor subfamily 5, group A, Key regulator of Cyp7a expression in liver. Linked to a variety of processes, such as bile acid member 2 metabolism and reverse cholesterol transport. Mbtps1 membrane-bound transcription factor Catalyzes the first step in the proteolytic activation of the Srebf proteins. peptidase, site 1 Srebf2 sterol regulatory element binding Transcription factor that controls cholesterol homeostasis by transcribing sterol-regulated genes. transcription factor 2 Gene Gene Name Function Symbol Bile acid metabolism Akr1c6 aldo-keto reductase family 1, member C1 May have a role in the transport and intrahepatic concentration of bile acids. Akr1d1 aldo-keto reductase family 1, member D1 Reduces BA intermediates 7-α,12-α-dihydroxy-4-cholesten-3-one & 7-α-hydroxy-4-cholesten-3-one. Amacr alpha-methylacyl-CoA racemase Responsible for the conversion of pristanoyl-CoA and C27-bile acyl-CoAs to their (S)-stereoisomers. Baat (Bat) bile acid CoA: amino acid N- Conjugates C24 bile acids to glycine or taurine before excretion into bile canaliculi.