Metabolic Network Analysis Predicts Efficacy of FDA-Approved Drugs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae
Journal of Fungi Article Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae Mohammad Sayari 1,2,*, Magrieta A. van der Nest 1,3, Emma T. Steenkamp 1, Saleh Rahimlou 4 , Almuth Hammerbacher 1 and Brenda D. Wingfield 1 1 Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (A.H.); brenda.wingfi[email protected] (B.D.W.) 2 Department of Plant Science, University of Manitoba, 222 Agriculture Building, Winnipeg, MB R3T 2N2, Canada 3 Biotechnology Platform, Agricultural Research Council (ARC), Onderstepoort Campus, Pretoria 0110, South Africa 4 Department of Mycology and Microbiology, University of Tartu, 14A Ravila, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Fax: +1-204-474-7528 Abstract: Terpenes represent the biggest group of natural compounds on earth. This large class of organic hydrocarbons is distributed among all cellular organisms, including fungi. The different classes of terpenes produced by fungi are mono, sesqui, di- and triterpenes, although triterpene ergosterol is the main sterol identified in cell membranes of these organisms. The availability of genomic data from members in the Ceratocystidaceae enabled the detection and characterization of the genes encoding the enzymes in the mevalonate and ergosterol biosynthetic pathways. Using Citation: Sayari, M.; van der Nest, a bioinformatics approach, fungal orthologs of sterol biosynthesis genes in nine different species M.A.; Steenkamp, E.T.; Rahimlou, S.; of the Ceratocystidaceae were identified. -

The Impact of Transcription on Metabolism in Prostate and Breast Cancers
25 9 Endocrine-Related N Poulose et al. From hormones to fats and 25:9 R435–R452 Cancer back REVIEW The impact of transcription on metabolism in prostate and breast cancers Ninu Poulose1, Ian G Mills1,2,* and Rebecca E Steele1,* 1Centre for Cancer Research and Cell Biology, Queen’s University of Belfast, Belfast, UK 2Nuffield Department of Surgical Sciences, John Radcliffe Hospital, University of Oxford, Oxford, UK Correspondence should be addressed to I G Mills: [email protected] *(I G Mills and R E Steele contributed equally to this work) Abstract Metabolic dysregulation is regarded as an important driver in cancer development and Key Words progression. The impact of transcriptional changes on metabolism has been intensively f androgen studied in hormone-dependent cancers, and in particular, in prostate and breast cancer. f androgen receptor These cancers have strong similarities in the function of important transcriptional f breast drivers, such as the oestrogen and androgen receptors, at the level of dietary risk and f oestrogen epidemiology, genetics and therapeutically. In this review, we will focus on the function f endocrine therapy of these nuclear hormone receptors and their downstream impact on metabolism, with a resistance particular focus on lipid metabolism. We go on to discuss how lipid metabolism remains dysregulated as the cancers progress. We conclude by discussing the opportunities that this presents for drug repurposing, imaging and the development and testing of new Endocrine-Related Cancer therapeutics and treatment combinations. (2018) 25, R435–R452 Introduction: prostate and breast cancer Sex hormones act through nuclear hormone receptors Early-stage PCa is dependent on androgens for survival and induce distinct transcriptional programmes essential and can be treated by androgen deprivation therapy; to male and female physiology. -

Halfanâ Brand of Halofantrine Hydrochloride Tablets
HL:L4 PRESCRIBING INFORMATION HALFANâ brand of halofantrine hydrochloride Tablets WARNING: HALFAN HAS BEEN SHOWN TO PROLONG QTc INTERVAL AT THE RECOMMENDED THERAPEUTIC DOSE. THERE HAVE BEEN RARE REPORTS OF SERIOUS VENTRICULAR DYSRHYTHMIAS SOMETIMES ASSOCIATED WITH DEATH, WHICH MAY BE SUDDEN. HALFAN IS THEREFORE NOT RECOMMENDED FOR USE IN COMBINATION WITH DRUGS OR CLINICAL CONDITIONS KNOWN TO PROLONG QTc INTERVAL, OR IN PATIENTS WHO HAVE PREVIOUSLY RECEIVED MEFLOQUINE, OR IN PATIENTS WITH KNOWN OR SUSPECTED VENTRICULAR DYSRHYTHMIAS, A-V CONDUCTION DISORDERS OR UNEXPLAINED SYNCOPAL ATTACKS. HALFAN SHOULD BE PRESCRIBED ONLY BY PHYSICIANS WHO HAVE SPECIAL COMPETENCE IN THE DIAGNOSIS AND TREATMENT OF MALARIA, AND WHO ARE EXPERIENCED IN THE USE OF ANTIMALARIAL DRUGS. PHYSICIANS SHOULD THOROUGHLY FAMILIARIZE THEMSELVES WITH THE COMPLETE CONTENTS OF THIS LEAFLET BEFORE PRESCRIBING HALFAN. DESCRIPTION Halfan (halofantrine hydrochloride) is an antimalarial drug available as tablets containing 250 mg of halofantrine hydrochloride (equivalent to 233 mg of the free base) for oral administration. The chemical name of halofantrine hydrochloride is 1,3-dichloro-a-[2-(dibutylamino) ethyl]- 6-(trifluoromethyl)-9-phenanthrene-methanol hydrochloride. The drug, a white to off-white crystalline compound, is practically insoluble in water. Halofantrine hydrochloride has a calculated molecular weight of 536.89. The empirical formula is C26H30Cl2F3NOHCl and the structural formula is 1 Inactive Ingredients Inactive ingredients are magnesium stearate, microcrystalline cellulose, povidone, pregelatinized starch, sodium starch glycolate and talc. CLINICAL PHARMACOLOGY The interindividual variability in the pharmacokinetic parameters of halofantrine is very wide and has led to great difficulty in precisely determining the pharmacokinetic characteristics of this product. Following administration of halofantrine hydrochloride tablets in single oral doses of 250 mg to 1000 mg to healthy volunteers, peak plasma levels were reached in 5 to 7 hours. -

33 34 35 Lipid Synthesis Laptop
BI/CH 422/622 Liver cytosol ANABOLISM OUTLINE: Photosynthesis Carbohydrate Biosynthesis in Animals Biosynthesis of Fatty Acids and Lipids Fatty Acids Triacylglycerides contrasts Membrane lipids location & transport Glycerophospholipids Synthesis Sphingolipids acetyl-CoA carboxylase Isoprene lipids: fatty acid synthase Ketone Bodies ACP priming 4 steps Cholesterol Control of fatty acid metabolism isoprene synth. ACC Joining Reciprocal control of b-ox Cholesterol Synth. Diversification of fatty acids Fates Eicosanoids Cholesterol esters Bile acids Prostaglandins,Thromboxanes, Steroid Hormones and Leukotrienes Metabolism & transport Control ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Synthesis Degradation Ketone body Utilization Isoprene Biosynthesis 1 Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates are transferred stepwise from ATP to mevalonate. • A third phosphate from ATP is added at the hydroxyl, followed by decarboxylation and elimination catalyzed by pyrophospho- mevalonate decarboxylase creates a pyrophosphorylated 5-C product: D3-isopentyl pyrophosphate (IPP) (isoprene). • Isomerization to a second isoprene dimethylallylpyrophosphate (DMAPP) gives two activated isoprene IPP compounds that act as precursors for D3-isopentyl pyrophosphate Isopentyl-D-pyrophosphate all of the other lipids in this class isomerase DMAPP Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates -

Update on Pathogenesis of Primary
177:3 C E Flück Update primary 177:3 R99–R111 Review adrenal insufficiency MECHANISMS IN ENDOCRINOLOGY Update on pathogenesis of primary adrenal insufficiency: beyond steroid enzyme deficiency and autoimmune adrenal destruction Correspondence Christa E Flück should be addressed Departments of Pediatrics and Clinical Research, Bern University Children’s Hospital Inselspital, University of Bern, to C E Flück Bern, Switzerland Email [email protected] Abstract Primary adrenal insufficiency (PAI) is potentially life threatening, but rare. In children, genetic defects prevail whereas adults suffer more often from acquired forms of PAI. The spectrum of genetic defects has increased in recent years with the use of next-generation sequencing methods and now has reached far beyond genetic defects in all known enzymes of adrenal steroidogenesis. Cofactor disorders such as P450 oxidoreductase (POR) deficiency manifesting as a complex form of congenital adrenal hyperplasia with a broad clinical phenotype have come to the fore. In patients with isolated familial glucocorticoid deficiency (FGD), in which no mutations in the genes for the ACTH receptor (MC2R) or its accessory protein MRAP have been found, non-classic steroidogenic acute regulatory protein (StAR) and CYP11A1 mutations have been described; and more recently novel mutations in genes such as nicotinamide nucleotide transhydrogenase (NNT) and thioredoxin reductase 2 (TRXR2) involved in the maintenance of the mitochondrial redox potential and generation of NADPH important for steroidogenesis and ROS detoxication have been discovered. European Journal European of Endocrinology In addition, whole exome sequencing approach also solved the genetics of some syndromic forms of PAI including IMAGe syndrome (CDKN1C), Irish traveler syndrome (MCM4), MIRAGE syndrome (SAMD9); and most recently a syndrome combining FGD with steroid-resistant nephrotic syndrome and ichthyosis caused by mutations in the gene for sphingosine-1-phosphate lyase 1 (SGPL1). -

A Key Mammalian Cholesterol Synthesis Enzyme, Squalene Monooxygenase, Is Allosterically Stabilized by Its Substrate
A key mammalian cholesterol synthesis enzyme, squalene monooxygenase, is allosterically stabilized by its substrate Hiromasa Yoshiokaa, Hudson W. Coatesb, Ngee Kiat Chuab, Yuichi Hashimotoa, Andrew J. Brownb,1, and Kenji Ohganea,c,1 aInstitute for Quantitative Biosciences, The University of Tokyo, 113-0032 Tokyo, Japan; bSchool of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW 2052, Australia; and cSynthetic Organic Chemistry Laboratory, RIKEN, 351-0198 Saitama, Japan Edited by Brenda A. Schulman, Max Planck Institute of Biochemistry, Martinsried, Germany, and approved February 18, 2020 (received for review September 20, 2019) Cholesterol biosynthesis is a high-cost process and, therefore, is the cognate E3 ligase for SM (9, 10), catalyzing its ubiquitination tightly regulated by both transcriptional and posttranslational in the N100 region (11). negative feedback mechanisms in response to the level of cellular Despite its importance in cholesterol-accelerated degradation, cholesterol. Squalene monooxygenase (SM, also known as squalene the structure and function of the SM-N100 region has not been epoxidase or SQLE) is a rate-limiting enzyme in the cholesterol fully resolved. Thus far, biochemical analyses have revealed the biosynthetic pathway and catalyzes epoxidation of squalene. The presence of a reentrant loop anchoring SM to the ER membrane stability of SM is negatively regulated by cholesterol via its and an amphipathic helix required for cholesterol-accelerated N-terminal regulatory domain (SM-N100). In this study, using a degradation (7, 8), while X-ray crystallography has revealed the SM-luciferase fusion reporter cell line, we performed a chemical architecture of the catalytic domain of SM (12). However, the genetics screen that identified inhibitors of SM itself as up-regulators highly hydrophobic and disordered nature of the N100 region of SM. -

Halofantrine and Primaquine for Radical Cure of Malaria in Irian Jaya, Indonesia D
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Public Health Resources Public Health Resources 1997 Halofantrine and primaquine for radical cure of malaria in Irian Jaya, Indonesia D. J. Fryauff U.S. NAMRU-2 (Jakarta), Box 3, APO AP 96520-8132, U.S.A. J. Kevin Baird U.S. NAMRU-2 (Jakarta), Box 3, APO AP 96520-8132, U.S.A., [email protected] H. Basri U.S. NAMRU-2 (Jakarta), Box 3, APO AP 96520-8132, U.S.A. I. Wiady U.S. NAMRU-2 (Jakarta), Box 3, APO AP 96520-8132, U.S.A. U.S. NAMRU-2 (Jakarta), Box 3, APO AP 96520-8132, U.S.A. See next page for additional authors Follow this and additional works at: http://digitalcommons.unl.edu/publichealthresources Fryauff, D. J.; Baird, J. Kevin; Basri, H.; Wiady, I.; U.S. NAMRU-2 (Jakarta), Box 3, APO AP 96520-8132, U.S.A.; Bangs, M. J.; Subianto, B.; Harjosuwarno, S.; Tjitra, E.; Richie, T. L.; and Hoffman, S. L., "Halofantrine and primaquine for radical cure of malaria in Irian Jaya, Indonesia" (1997). Public Health Resources. 376. http://digitalcommons.unl.edu/publichealthresources/376 This Article is brought to you for free and open access by the Public Health Resources at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Public Health Resources by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors D. J. Fryauff; J. Kevin Baird; H. Basri; I. Wiady; U.S. NAMRU-2 (Jakarta), Box 3, APO AP 96520-8132, U.S.A.; M. J. -

Integrating Data Mining, Network Pharmacology, and Molecular
Integrating Data Mining, Network Pharmacology, and Molecular Docking Verication to Investigate the Molecular Mechanism of Traditional Chinese Medicine Prescriptions for Treating Male Infertility Xue Bai Beijing University of Chinese Medicine Yibo Tang Beijing University of Chinese Medicine Qiang Li Beijing University of Chinese Medicine Guimin Liu Beijing University of Chinese Medicine Dan Liu Beijing University of Chinese Medicine Xiaolei Fan Beijing University of Chinese Medicine Zhejun Liu Beijing University of Chinese Medicine Shujun Yu Beijing University of Chinese Medicine Tian Tang Beijing University of Chinese Medicine Shuyan Wang Beijing University of Chinese Medicine Lingru Li Beijing University of Chinese Medicine Kailin Zhou Beijing University of Chinese Medicine Yanfei Zheng Beijing University of Chinese Medicine Zhenquan Liu ( [email protected] ) Research Page 1/42 Keywords: Traditional Chinese medicine, male infertility, data mining, network pharmacology, molecular docking Posted Date: March 4th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-264555/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/42 Abstract Background: Male infertility (MI) affects almost 5% adult men worldwide, and 75% of these cases are unexplained idiopathic. There are limitations in the current treatment due to the unclear mechanism of MI, which highlight the urgent need for a more effective strategy or drug. Traditional Chinese Medicine (TCM) prescriptions have been used to treat MI for thousands of years, but their molecular mechanism is not well dened. Methods: Aiming at revealing the molecular mechanism of TCM prescriptions on MI, a comprehensive strategy integrating data mining, network pharmacology, and molecular docking verication was performed. -

(Artemether / Lumefantrine 20Mg/120 Mg) Tablets
SUMMARY OF PRODUCT CHARACTERISTICS Page 1 of 13 1. NAME OF THE MEDICINAL PRODUCT Lumartem (Artemether / Lumefantrine 20mg/120 mg) Tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each tablet contains: 20 mg artemether and 120 mg lumefantrine. For a full list of excipients see section 6.1. 3. PHARMACEUTICAL FORM Yellow coloured, circular, uncoated, flat faced, bevelled edged, matt finished tablets with a break line on one side and plain on the other side. The scoreline is only to facilitate breaking for ease of swallowing and not to divide into equal doses. 4. CLINICAL PARTICULARS 4.1 Therapeutic indication Artemether 20 mg and Lumefantrine 120 mg Tablets is indicated for the treatment of uncomplicated cases of malaria due to Plasmodium falciparum in adults, children and infants of 5 kg and above. The most recent official guidelines on the appropriate use of antimalarial agents and local information on the prevalence of resistance to antimalarial drugs must be taken into consideration for deciding on the appropriateness of therapy with Artemether 20 mg and Lumefantrine 120 mg Tablets. Official guidance will normally include WHO (http://whqlibdoc.who.int/publications/2010/9789241547925_eng.pdf) and local health authorities’ guidelines (see also sections 4.4 and 5.1). 4.2 Posology and method of administration Tablets for oral administration. Page 2 of 13 Number of Artemether 20 mg and Lumefantrine 120 mg Tablets for treatment according to weight bands st nd rd Weight range 1 day of 2 day of 3 day treatment treatment of treatment ≥ 5kg -
Figures Intermediary Ontogeny.Pptx
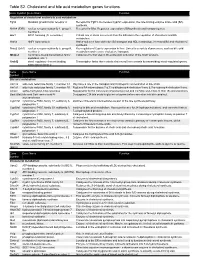
Table S2. Cholesterol and bile acid metabolism genes functions. Gene Symbol Gene Name Function Regulation of cholesterol and/or bile acid metabolism Fgfr4 fibroblast growth factor receptor 4 Receptor for Fgf15. Decreases Cyp7a1 expression, the rate-limiting enzyme in bile acid (BA) synthesis. Nr1h4 (FXR) nuclear receptor subfamily 1, group H, Receptor for BAs. Regulates expression of BAsynthesis and transport genes. member 4 Arv1 ARV1 homolog (S. cerevisiae) Critical role in sterol movement from the ER and in the regulation of cholesterol and BA metabolism. Hnf1a HNF1 homeobox A Hnf1a-null mice have defective BA transport and HDL metabolism, increased BA and cholesterol synthesis. Nr5a2 (Lrh1) nuclear receptor subfamily 5, group A, Key regulator of Cyp7a expression in liver. Linked to a variety of processes, such as bile acid member 2 metabolism and reverse cholesterol transport. Mbtps1 membrane-bound transcription factor Catalyzes the first step in the proteolytic activation of the Srebf proteins. peptidase, site 1 Srebf2 sterol regulatory element binding Transcription factor that controls cholesterol homeostasis by transcribing sterol-regulated genes. transcription factor 2 Gene Gene Name Function Symbol Bile acid metabolism Akr1c6 aldo-keto reductase family 1, member C1 May have a role in the transport and intrahepatic concentration of bile acids. Akr1d1 aldo-keto reductase family 1, member D1 Reduces BA intermediates 7-α,12-α-dihydroxy-4-cholesten-3-one & 7-α-hydroxy-4-cholesten-3-one. Amacr alpha-methylacyl-CoA racemase Responsible for the conversion of pristanoyl-CoA and C27-bile acyl-CoAs to their (S)-stereoisomers. Baat (Bat) bile acid CoA: amino acid N- Conjugates C24 bile acids to glycine or taurine before excretion into bile canaliculi. -

Open Full Page
Published OnlineFirst May 7, 2019; DOI: 10.1158/0008-5472.CAN-18-2529 Cancer Molecular Cell Biology Research Upregulation of Scavenger Receptor B1 Is Required for Steroidogenic and Nonsteroidogenic Cholesterol Metabolism in Prostate Cancer Jacob A. Gordon1,2, Jake W. Noble1, Ankur Midha3, Fatemeh Derakhshan4, Gang Wang4, Hans H. Adomat1, Emma S. Tomlinson Guns1,2, Yen-Yi Lin1, Shancheng Ren5, Collin C. Collins1, Peter S. Nelson6, Colm Morrissey7, Kishor M. Wasan2,8, and Michael E. Cox1,2,9 Abstract Aberrant cholesterol metabolism is increasingly appre- viability and induced endoplasmic reticulum stress and ciated to be essential for prostate cancer initiation and autophagy. The inability of exogenous steroids to reverse progression. Transcript expression of the high-density lipo- these effects indicates that AR pathway activation is insuffi- protein-cholesterol receptor scavenger receptor B1 (SR-B1) cient to overcome cytotoxic stress caused by a decrease in the is elevated in primary prostate cancer. Hypothesizing that availability of cholesterol. Furthermore, SR-B1 antagonism SR-B1 expression may help facilitate malignant transforma- decreased cholesterol uptake, growth, and viability of the tion, we document increased SR-B1 protein and transcript AR-null CRPC cell model PC-3, and the small-molecule SR- expression in prostate cancer relative to normal prostate B1 antagonist block lipid transport-1 decreased xenograft epithelium that persists in lethal castration-resistant prostate growth rate despite poor pharmacologic properties. Overall, cancer (CRPC) metastasis. As intratumoral steroid synthesis our findings show that SR-B1 is upregulated in primary and from the precursor cholesterol can drive androgen receptor castration-resistant disease and is essential for cholesterol (AR) pathway activity in CRPC, we screened androgenic uptake needed to drive both steroidogenic and nonsteroi- benign and cancer cell lines for sensitivity to SR-B1 antag- dogenic biogenic pathways, thus implicating SR-B1 as a onism. -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc.