Genome Sequence of the Deep-Sea -Proteobacterium Idiomarina
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Supplemental Methods
Supplemental Methods: Sample Collection Duplicate surface samples were collected from the Amazon River plume aboard the R/V Knorr in June 2010 (4 52.71’N, 51 21.59’W) during a period of high river discharge. The collection site (Station 10, 4° 52.71’N, 51° 21.59’W; S = 21.0; T = 29.6°C), located ~ 500 Km to the north of the Amazon River mouth, was characterized by the presence of coastal diatoms in the top 8 m of the water column. Sampling was conducted between 0700 and 0900 local time by gently impeller pumping (modified Rule 1800 submersible sump pump) surface water through 10 m of tygon tubing (3 cm) to the ship's deck where it then flowed through a 156 µm mesh into 20 L carboys. In the lab, cells were partitioned into two size fractions by sequential filtration (using a Masterflex peristaltic pump) of the pre-filtered seawater through a 2.0 µm pore-size, 142 mm diameter polycarbonate (PCTE) membrane filter (Sterlitech Corporation, Kent, CWA) and a 0.22 µm pore-size, 142 mm diameter Supor membrane filter (Pall, Port Washington, NY). Metagenomic and non-selective metatranscriptomic analyses were conducted on both pore-size filters; poly(A)-selected (eukaryote-dominated) metatranscriptomic analyses were conducted only on the larger pore-size filter (2.0 µm pore-size). All filters were immediately submerged in RNAlater (Applied Biosystems, Austin, TX) in sterile 50 mL conical tubes, incubated at room temperature overnight and then stored at -80oC until extraction. Filtration and stabilization of each sample was completed within 30 min of water collection. -

Computational and Systems Biology Themed Issue
View Article Online / Journal Homepage / Table of Contents for this issue Molecular BioSystems This article was published as part of the Computational and Systems Biology themed issue Please take a look at the full table of contents to access the other papers in this issue. Open Access Article. Published on 13 October 2009. Downloaded 9/23/2021 6:41:00 PM. View Article Online PAPER www.rsc.org/molecularbiosystems | Molecular BioSystems Amidoligases with ATP-grasp, glutamine synthetase-like and acetyltransferase-like domains: synthesis of novel metabolites and peptide modifications of proteinswz Lakshminarayan M. Iyer,a Saraswathi Abhiman,a A. Maxwell Burroughsb and L. Aravind*a Received 28th August 2009, Accepted 28th August 2009 First published as an Advance Article on the web 13th October 2009 DOI: 10.1039/b917682a Recent studies have shown that the ubiquitin system had its origins in ancient cofactor/amino acid biosynthesis pathways. Preliminary studies also indicated that conjugation systems for other peptide tags on proteins, such as pupylation, have evolutionary links to cofactor/amino acid biosynthesis pathways. Following up on these observations, we systematically investigated the non-ribosomal amidoligases of the ATP-grasp, glutamine synthetase-like and acetyltransferase folds by classifying the known members and identifying novel versions. We then established their contextual connections using information from domain architectures and conserved gene neighborhoods. This showed remarkable, previously uncharacterized functional links between diverse peptide ligases, several peptidases of unrelated folds and enzymes involved in synthesis of modified amino acids. Using the network of contextual connections we were able to predict numerous novel pathways for peptide synthesis and modification, amine-utilization, secondary metabolite synthesis and potential peptide-tagging systems. -

Comparative Genomics of the Mycobacterium Ulcerans And
MPhil Thesis Ken Doig (197821835) MPhil Thesis – 2012 Candidate Kenneth Douglas Doig Student No. 197821835 Title Comparative genomics of the Mycobacterium ulcerans and Mycobacterium marinum complex Status Submitted in total fulfilment of the requirements of the degree of Master of Philosophy Date September 2012 Supervisors A/Prof. Tim Stinear1 Dr. Torsten Seemann2 Prof. Richard Strugnell1 Department Microbiology and Immunology Faculty Medicine, Dentistry and Health Sciences The University of Melbourne 1. Department of Microbiology and Immunology, University of Melbourne, Parkville, Australia 2. Victorian Bioinformatics Consortium, Monash University, Clayton, Australia MPhil_Thesis-22.doc Page 1 of 1 MPhil Thesis Ken Doig (197821835) Abstract Buruli ulcer is a neglected tropical disease which is prevalent in Western Africa. Its etiological agent, Mycobacteria ulcerans occurs in a wide range of hosts and environments across the world. In contrast to its progenitor Mycobacteria marinum, the bacteria and related strains produce a toxin mycolactone, an immunosuppressive polyketide which gives rise to its pathogenicity. Bacterial pathogenesis has a number of hallmarks such as; an evolutionary bottleneck, insertion sequence (IS) expansion, pseudogene increase, genome reduction, horizontal gene transfers (HGT) and adaption to a new niche. M. ulcerans shows all of these characteristics and is a fine model for gaining a deeper understanding of the mechanics of bacterial pathogenesis in general and specifically for M. ulcerans and related strains. This research analyses the genomic makeup of 35 isolates that produce mycolactone and five M. marinum isolates. Such analysis have recently become possible through the rapid technological development of high throughput sequencing (HTS) allowing whole genome sequences of all isolates to be compared and contrasted. -

Table S3. Bootstrapped Frequency of COG Occurrences for Specific Metabolic Processes Encoded in the Antarctic Bacterioplankton Environmental Genomes
Table S3. Bootstrapped frequency of COG occurrences for specific metabolic processes encoded in the Antarctic bacterioplankton environmental genomes. Category COG Predicted Protein WEG SEG Inorganic carbon COG1850 Ribulose 1,5-bisphosphate carboxylase, large subunit 4.34 0.85 COG4451 Ribulose bisphosphate carboxylase small subunit 1.19 0.83 COG0574 Phosphoenolpyruvate synthase/pyruvate phosphate dikinase 13.7 6.63 COG4770 Acetyl/propionyl-CoA carboxylase, alpha subunit 7.91 4.85 Sulfur Metabolism (inorganic) COG2897 Rhodanese-related sulfurtransferase 5.64 3.25 COG0607 Rhodanese-related sulfurtransferase 6.02 5.68 COG2895 GTPases - Sulfate adenylate transferase subunit 1 2.28 4.88 COG1054 Predicted sulfurtransferase 2.53 4.17 COG0306 Phosphate/sulphate permeases 7.56 3.27 COG0659 Sulfate permease and related transporters (MFS superfamily) 15.72 15.22 COG0529 Adenylylsulfate kinase and related kinases 1.98 4.16 COG0225 Peptide methionine sulfoxide reductase 7.03 3.22 COG0229 Conserved domain frequently associated with peptide methionine sulfoxide reductase 3.77 2.47 COG0641 Arylsulfatase regulator (Fe-S oxidoreductase) 0.67 0 COG0526 Thiol-disulfide isomerase and thioredoxins 6.99 4.12 COG1651 Protein-disulfide isomerase 10.75 1.65 COG2041 Sulfite oxidase and related enzymes 5.51 3.05 COG2046 ATP sulfurylase (sulfate adenylyltransferase) 4.34 3.46 COG2221 Dissimilatory sulfite reductase (desulfoviridin), alpha and beta subunits 1.21 0 COG2920 Dissimilatory sulfite reductase (desulfoviridin), gamma subunit 1.9 0 COG0155 Sulfite reductase, -

ABSTRACT BOZDAG, AHMET. Investigation of Methanol and Formaldehyde Metabolism in Bacillus Methanolicus
ABSTRACT BOZDAG, AHMET. Investigation of Methanol and Formaldehyde Metabolism in Bacillus methanolicus.(Under the direction of Prof. Michael C. Flickinger). Bacillus methanolicus is a Gram-positive aerobic methylotroph growing optimally at 50-53 °C. Wild-type strains of B. methanolicus have been reported to secrete 58 g/l of L- glutamate in fed-batch cultures. Mutants of B. methanolicus created via classical mutangenesis can secrete 37 g/l of L-lysine, at 50 °C. The genes required for methylotrophyin B. methanolicus are encoded by an endogenous plasmid, pBM19 in strain MGA3, except for hexulose phosphate synthase (hps) and phosphohexuloisomerase (phi) which are encoded on the chromosome.It is a promising candidate for industrial production of chemical intermediates or amino acids from methanol. B. methanolicus employs the ribulose monophospate (RuMP) pathway to assimilate the carbon derived from the methanol, but enzymes that dissimilate carbon are not identified, although formaldehyde and formate were identified as intermediates by 13C NMR. It is important to understand how methanol is oxidized to formaldehyde and then, to formate and carbon dioxide. This study aims to elucidate the methanol dissimilation pathway of B. methanolicus. Growth rates of B. methanolicus MGA3 were assessed on methanol, mannitol, and glucose as a substrate. B. methanolicus achieved maximum growth rate, µmax, when growing on 25 mM methanol, 0.65±0.007 h-1, and it gradually decreased to 0.231±0.004 h-1 at 2Mmethanol concentration which demonstrates substrate inhibition. The maximum growth rates (µmax) of B. methanolicus MGA3 on mannitol and glucose are 0.532±0.002 and 0.336±0.003 h-1, respectively. -

Planctomycetes Attached to Algal Surfaces: Insight Into Their Genomes
MSc 2.º CICLO FCUP 2015 into Planctomycetes their Planctomycetes genomes attached attached to algal surfaces: Insight into to algal their genomes surfaces Mafalda Seabra Faria : Insight : Dissertação de Mestrado apresentada à Faculdade de Ciências da Universidade do Porto Laboratório de Ecofisiologia Microbiana da Universidade do Porto Biologia Celular e Molecular 2014/2015 Mafalda Seabra Faria Seabra Mafalda I FCUP Planctomycetes attached to algal surfaces: Insight into their genomes Planctomycetes attached to algal surfaces: Insight into their genomes Mafalda Seabra Faria Mestrado em Biologia Celular e Molecular Biologia 2015 Orientador Olga Maria Oliveira da Silva Lage, Professora Auxiliar, Faculdade de Ciências da Universidade do Porto Co-orientador Jens Harder, Senior Scientist and Professor, Max Planck Institute for Marine Microbiology FCUP II Planctomycetes attached to algal surfaces: Insight into their genomes Todas as correções determinadas pelo júri, e só essas, foram efetuadas. O Presidente do Júri, Porto, ______/______/_________ FCUP III Planctomycetes attached to algal surfaces: Insight into their genomes FCUP IV Planctomycetes attached to algal surfaces: Insight into their genomes “Tell me and I forget, teach me and I may remember, involve me and I learn.” Benjamin Franklin FCUP V Planctomycetes attached to algal surfaces: Insight into their genomes FCUP VI Planctomycetes attached to algal surfaces: Insight into their genomes Acknowledgements Foremost, I would like to express my sincere gratitude to my supervisor Professor -
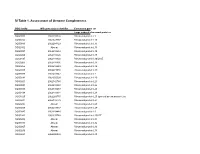
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Yixing Zhang
CORE Metadata, citation and similar papers at core.ac.uk Provided by K-State Research Exchange PRODUCTION OF NITROGEN-BASED PLATFORM CHEMICAL: CYANOPHYCIN BIOSYNTHESIS USING RECOMBINANT ESCHERICHIA COLI by YIXING ZHANG B.Sc., China Agricultural University, 2008 A THESIS Submitted in partial fulfillment of the requirements for the degree MASTER OF SCIENCE Department of Grain Science and Industry College of Agriculture KANSAS STATE UNIVERSITY Manhattan, Kansas 2010 Approved by: Major Professor Praveen V. Vadlani Abstract Synthesis of chemical derivatives from finite fossil fuels requires considerable energy inputs and leaves an undesirable environmental footprint. The emerging biorefinery approach leads to sustainable processing of biomass into a wide spectrum of bio-based products, catering to food, feed, chemicals, materials, and bioenergy industries. Cyanophycin (multi-L-arginyl-poly-L-aspartic acid, CGP) is a non-ribosomally synthesized reserve polypeptide, which consists of equimolar amounts of arginine and aspartic acid arranged as a polyaspartate backbone and arginine as the side chain. Cyanophycin is a source of the constituent N-functionalized platform chemical, which can be further processed into many other chemicals of importance. It can be hydrolyzed in mild condition to its constituent amino acids - aspartic acid and arginine. These amino acids may be utilized directly in food and pharmaceutical applications. Based on the chemical structure of these amino acids and the presence of functionalized nitrogen-containing groups, it is conceivable that a number of industrial chemicals can be synthesized, for example: 1, 4-butanediamine, a co-monomer in the production of nylon-4, 6. Other chemicals which could be obtained from cyanophcyin, that are currently prepared from fossil resources, include 1,4-butanediol and urea. -

Supporting Information
Electronic Supplementary Material (ESI) for Metallomics. This journal is © The Royal Society of Chemistry 2019 Supporting information for Metabolomic and proteomic changes induced by growth inhibitory concentrations of copper in the biofilm-forming marine bacterium Pseudoalteromonas lipolytica Laurie Favre,a Annick Ortalo-Magné,a,* Lionel Kerloch,a Carole Pichereaux,b,c Benjamin Misson,d Jean-François Brianda, Cédric Garnierd and Gérald Culiolia,* a Université de Toulon, MAPIEM EA 4323, Toulon, France. b Fédération de Recherche FR3450, Agrobiosciences, Interaction et Biodiversité (AIB), CNRS, Toulouse, France.. c Institut de Pharmacologie et de Biologie Structurale, IPBS, Université de Toulouse, CNRS, UPS, Toulouse, France. d Univ de Toulon, Aix Marseille Univ, CNRS, IRD, MIO UM 110, Mediterranean Institute of Oceanography, La Garde, France. Detailed description of the experimental protocol used for Cu chemical speciation. Differential pulse anodic striping voltammetry (DPASV) measurements were performed to assess Cu chemical speciation in both VNSS and MB medium. All voltammetric measurements were performed using a PGSTAT12 potentiostat (EcoChemie, The Netherlands) equipped with a Metrohm 663 VA stand (Metrohm, Switzerland) on an Hg drop electrode. Analysis of the voltammetric peaks' (after 2nd derivative transformation) and construction of the pseudo- polarograms were performed automatically using ECDSoft software. The experimental procedure adapted from Nicolau et al. (2008) and Bravin et al. (2012) is explained briefly below. Pseudo-polarographic measurements (DPASV measurement with Edep from -1.0 to 0.0V by steps of 100 mV, using a deposition time of 30s) were initially performed on UV-irradiated seawater (collected outside the Toulon bay: ambient Cu concentration of 2.5 nM) at pH < 2 (by adding HNO3 Suprapur grade, Merck), to determine the deposition potential (Edep -0.35V) corresponding to the electrochemically "labile" Cu fraction (sum of free Cu ions plus inorganically bound Cu) (Figure S2). -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000091785Cyc: Legionella longbeachae NSW150 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari sn-glycerol spermidine 3-phosphate putrescine a sulfonate nitrate hydrogencarbonate predicted predicted ABC ABC transporter transporter RS16610 of sn- of spermidine/ glycerol 3- putrescine phosphate a sulfonate nitrate spermidine hydrogencarbonate putrescine sn-glycerol 3-phosphate Amino Acid Degradation Amine and Polyamine Biosynthesis Hormone Biosynthesis Aldehyde Degradation glutaminyl-tRNA gln Aminoacyl-tRNA Charging N 6 -(3-methylbut- a 1-palmitoyl-2- L-alanyl-γ-D- L-alanyl-γ-D- a [bis(guanylyl a [protein] biosynthesis via transamidation core a peptide with homospermidine homospermidine indole-3-acetate L-leucine degradation I a sulfurated 2-en-1-yl)- acyl-sn-glycerol glutamyl-meso-2,6- glutamyl-meso- UDP-N-acetyl- a [glutamine- γ-L-glutamyl- Polyprenyl Biosynthesis L-proline degradation I L-tyrosine degradation I methylglyoxal degradation I oligosaccharide- ATP molybdopterin) cys a type IV prepilin gly an N-terminal C-terminal glu biosynthesis II L-arginine degradation [sulfur carrier] 37 synthetase]- ATP biosynthesis I biosynthesis TCA cycle TCA cycle I (prokaryotic) adenosine lipid A (E.