Computational and Systems Biology Themed Issue
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Supplemental Methods
Supplemental Methods: Sample Collection Duplicate surface samples were collected from the Amazon River plume aboard the R/V Knorr in June 2010 (4 52.71’N, 51 21.59’W) during a period of high river discharge. The collection site (Station 10, 4° 52.71’N, 51° 21.59’W; S = 21.0; T = 29.6°C), located ~ 500 Km to the north of the Amazon River mouth, was characterized by the presence of coastal diatoms in the top 8 m of the water column. Sampling was conducted between 0700 and 0900 local time by gently impeller pumping (modified Rule 1800 submersible sump pump) surface water through 10 m of tygon tubing (3 cm) to the ship's deck where it then flowed through a 156 µm mesh into 20 L carboys. In the lab, cells were partitioned into two size fractions by sequential filtration (using a Masterflex peristaltic pump) of the pre-filtered seawater through a 2.0 µm pore-size, 142 mm diameter polycarbonate (PCTE) membrane filter (Sterlitech Corporation, Kent, CWA) and a 0.22 µm pore-size, 142 mm diameter Supor membrane filter (Pall, Port Washington, NY). Metagenomic and non-selective metatranscriptomic analyses were conducted on both pore-size filters; poly(A)-selected (eukaryote-dominated) metatranscriptomic analyses were conducted only on the larger pore-size filter (2.0 µm pore-size). All filters were immediately submerged in RNAlater (Applied Biosystems, Austin, TX) in sterile 50 mL conical tubes, incubated at room temperature overnight and then stored at -80oC until extraction. Filtration and stabilization of each sample was completed within 30 min of water collection. -

Journal of Chromatography
aphy & S r ep og a t r a a t m i o o r n Lilla et al., J Chromatograph Separat Techniq 2012, 3:2 h T e C c f Journal of Chromatography h DOI: 10.4172/2157-7064.1000122 o n l i a q ISSN:n 2157-7064 u r e u s o J Separation Techniques Research Article OpenOpen Access Access Structural Characterization of Transglutaminase-Catalyzed Casein Cross- Linking Sergio Lilla1,2, Gianfranco Mamone2, Maria Adalgisa Nicolai1, Lina Chianese1, Gianluca Picariello2, Simonetta Caira2 and Francesco Addeo1,2* 1Dipartimento di Scienza degli Alimenti, University of Naples “Federico II”, Parco Gussone, Portici 80055, Italy 2Istituto di Scienze dell’Alimentazione (ISA) – CNR, Via Roma 64, 83100 Avellino, Italy Abstract Microbial transglutaminase is used in the food industry to improve texture by catalyzing protein cross-linking. Casein is a well-known transglutaminase substrate, but the complete role of glutamine (Q) and lysine (K) residues in its cross-linking is not fully understood. In this study, we describe the characterization of microbial Transglutaminase -modified casein using a combination of immunological and proteomic techniques. Using 5-(biotinamido)pentylamine as an acyl acceptor probe, three Q residues of β-casein and one of αs1-casein were found to participate as acyl donors. However, no Q-residues were involved in network formation with κ-casein or αs2-casein. Q and K residues in the ε-(γ-glutamyl)lysine-isopeptide bonds β-casein were identified by nanoelectrospray tandem mass spectrometry of the proteolytic digests. This work reports our progress toward a better understanding of the function and mechanism of action of microbial transglutaminase-mediated proteins. -

ABSTRACT Studies on Bovine Γ-Glutamylamine Cyclotransferase
ABSTRACT Studies on Bovine γ-Glutamylamine Cyclotransferase Maryuri Roca Mentor: Mary Lynn Trawick, Ph.D. The purification and study of proteins are cooperative processes because at least partially purified protein is needed in order to study its properties, and certain information about the protein’s properties is required in order to design its purification. Particularly difficult to purify is γ- glutamylamine cyclotransferase (γGACT ) which catalyzes the cyclization of the γ-glutamyl moiety in L-γ-glutamylamines, notably Nε−(γ-glutamyl)lysine. From this last activity the function of the enzyme is speculated to be related to the catabolism of transglutaminase products; although, there is no direct evidence of this. Electrophoretically pure bovine γGACT was obtained using preparative ultracentrifugation, anion exchange chromatography on DEAE-Sepharose, ammonium sulfate fractionation and precipitation, size exclusion chromatography on Sephacryl S100, anion exchange chromatography on Mono-Q under reducing conditions, isoelectric focusing of the alkylated sample, electroelution, electrophoresis, ultrafiltration, and lyophilization. The enzyme was purified more than 2,000 fold to a specific activity of more than 1,300U/mg of enzyme. A monomeric enzyme of molecular mass of 22,000 Daltons was observed. Anion exchange chromatography on a Mono Q GL column revealed two forms of the enzyme with pIs of 6.86 and 6.62 under non-reducing conditions, and a single form of pI 6.62 under reducing conditions. γGACT was then subjected to analytical isoelectric focusing and the active fraction appeared as a single band on SDS-PAGE. Amino acid sequencing of the tryptic digest of the band from SDS- PAGE corresponding to the enzyme was carried out by microcapillary reverse-phase HPLC nano-eletrospray tandem mass spectrometry; 42 proteins and protein fragments of similar mass and pI as that of γGACT were obtained. -

Amidoligases with ATP-Grasp, Glutamine Synthetase-Like and Acetyltransferase-Like Domains: Synthesis of Novel Metabolites and Peptide Modifications of Proteinswz
View Article Online / Journal Homepage / Table of Contents for this issue Molecular BioSystems This article was published as part of the Computational and Systems Biology themed issue Please take a look at the full table of contents to access the other papers in this issue. Open Access Article. Published on 13 October 2009. Downloaded 9/27/2021 9:23:51 AM. View Article Online PAPER www.rsc.org/molecularbiosystems | Molecular BioSystems Amidoligases with ATP-grasp, glutamine synthetase-like and acetyltransferase-like domains: synthesis of novel metabolites and peptide modifications of proteinswz Lakshminarayan M. Iyer,a Saraswathi Abhiman,a A. Maxwell Burroughsb and L. Aravind*a Received 28th August 2009, Accepted 28th August 2009 First published as an Advance Article on the web 13th October 2009 DOI: 10.1039/b917682a Recent studies have shown that the ubiquitin system had its origins in ancient cofactor/amino acid biosynthesis pathways. Preliminary studies also indicated that conjugation systems for other peptide tags on proteins, such as pupylation, have evolutionary links to cofactor/amino acid biosynthesis pathways. Following up on these observations, we systematically investigated the non-ribosomal amidoligases of the ATP-grasp, glutamine synthetase-like and acetyltransferase folds by classifying the known members and identifying novel versions. We then established their contextual connections using information from domain architectures and conserved gene neighborhoods. This showed remarkable, previously uncharacterized functional links between diverse peptide ligases, several peptidases of unrelated folds and enzymes involved in synthesis of modified amino acids. Using the network of contextual connections we were able to predict numerous novel pathways for peptide synthesis and modification, amine-utilization, secondary metabolite synthesis and potential peptide-tagging systems. -

Role of Transglutaminase 2 in Cell Death, Survival, and Fibrosis
cells Review Role of Transglutaminase 2 in Cell Death, Survival, and Fibrosis Hideki Tatsukawa * and Kiyotaka Hitomi Cellular Biochemistry Laboratory, Graduate School of Pharmaceutical Sciences, Nagoya University, Tokai National Higher Education and Research System, Nagoya 464-8601, Aichi, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-52-747-6808 Abstract: Transglutaminase 2 (TG2) is a ubiquitously expressed enzyme catalyzing the crosslink- ing between Gln and Lys residues and involved in various pathophysiological events. Besides this crosslinking activity, TG2 functions as a deamidase, GTPase, isopeptidase, adapter/scaffold, protein disulfide isomerase, and kinase. It also plays a role in the regulation of hypusination and serotonylation. Through these activities, TG2 is involved in cell growth, differentiation, cell death, inflammation, tissue repair, and fibrosis. Depending on the cell type and stimulus, TG2 changes its subcellular localization and biological activity, leading to cell death or survival. In normal unstressed cells, intracellular TG2 exhibits a GTP-bound closed conformation, exerting prosurvival functions. However, upon cell stimulation with Ca2+ or other factors, TG2 adopts a Ca2+-bound open confor- mation, demonstrating a transamidase activity involved in cell death or survival. These functional discrepancies of TG2 open form might be caused by its multifunctional nature, the existence of splicing variants, the cell type and stimulus, and the genetic backgrounds and variations of the mouse models used. TG2 is also involved in the phagocytosis of dead cells by macrophages and in fibrosis during tissue repair. Here, we summarize and discuss the multifunctional and controversial Citation: Tatsukawa, H.; Hitomi, K. roles of TG2, focusing on cell death/survival and fibrosis. -

1114 Tissue Transglutaminase (TG2) and Mitochondrial Function And
[Frontiers In Bioscience, Landmark, 22, 1114-1137, March 1, 2017] Tissue transglutaminase (TG2) and mitochondrial function and dysfunction Thung-S. Lai 1, Cheng-Jui Lin 2,3, Yu-Ting Wu4, Chih-Jen Wu2,5,6 1Institute of Biomedical Science, Mackay Medical College, New Taipei City, Taiwan, ROC, 2Nephrology/ Department of Internal Medicine, Mackay Memorial Hospital, Taipei, Taiwan, ROC, 3Nursing and Management, Mackay Junior College of Medicine, Taipei, Taiwan, ROC, 4Institute of Biochemistry and Molecular Biology, National Yang-Ming University, Taipei, Taiwan, 5Department of Medicine, Mackay Medical College, New Taipei City, Taiwan, ROC, 6Graduate Institute of Medical Science, Taipei Medical University, Taipei, Taiwan, ROC TABLE OF CONTENTS 1. Abstract 2. Introduction 3. TG2: a multifunctional enzyme. 3.1. Transamidation Reaction (TGase function) 3.1.1. Inter- or intra-molecular crosslinking 3.1.2. Aminylation 3.1.3. Deamidation 3.2. Isopeptidase activity 3.3. Protein Disulfide Isomerase (PDI) activity 3.4. GTP/ATP hydrolysis activity 4. Structure and function of TG2 4.1. TGase active site 4.2. GTP and ATP binding site 5. Regulation of in vivo TGase activity by GTP, redox, and nitric oxide (NO) 5.1. Regulation of in vivo TGase activity by GTP 5.2. Regulation of in vivo TGase activity by redox 5.3. Regulation of in vivo TGase activity by NO 6. Regulation of TG2 expression 6.1. NFkB regulates the expression of TG2 6.2. Hypoxia regulates the expression of TG2 6.3. TGFb regulates the expression of TG2 6.4. Oxidative stress and EGF up-regulate the expression of TG2. 7. TG2 is localized in mitochondria and several other locations 8. -

Comparative Genomics of the Mycobacterium Ulcerans And
MPhil Thesis Ken Doig (197821835) MPhil Thesis – 2012 Candidate Kenneth Douglas Doig Student No. 197821835 Title Comparative genomics of the Mycobacterium ulcerans and Mycobacterium marinum complex Status Submitted in total fulfilment of the requirements of the degree of Master of Philosophy Date September 2012 Supervisors A/Prof. Tim Stinear1 Dr. Torsten Seemann2 Prof. Richard Strugnell1 Department Microbiology and Immunology Faculty Medicine, Dentistry and Health Sciences The University of Melbourne 1. Department of Microbiology and Immunology, University of Melbourne, Parkville, Australia 2. Victorian Bioinformatics Consortium, Monash University, Clayton, Australia MPhil_Thesis-22.doc Page 1 of 1 MPhil Thesis Ken Doig (197821835) Abstract Buruli ulcer is a neglected tropical disease which is prevalent in Western Africa. Its etiological agent, Mycobacteria ulcerans occurs in a wide range of hosts and environments across the world. In contrast to its progenitor Mycobacteria marinum, the bacteria and related strains produce a toxin mycolactone, an immunosuppressive polyketide which gives rise to its pathogenicity. Bacterial pathogenesis has a number of hallmarks such as; an evolutionary bottleneck, insertion sequence (IS) expansion, pseudogene increase, genome reduction, horizontal gene transfers (HGT) and adaption to a new niche. M. ulcerans shows all of these characteristics and is a fine model for gaining a deeper understanding of the mechanics of bacterial pathogenesis in general and specifically for M. ulcerans and related strains. This research analyses the genomic makeup of 35 isolates that produce mycolactone and five M. marinum isolates. Such analysis have recently become possible through the rapid technological development of high throughput sequencing (HTS) allowing whole genome sequences of all isolates to be compared and contrasted. -

Table S3. Bootstrapped Frequency of COG Occurrences for Specific Metabolic Processes Encoded in the Antarctic Bacterioplankton Environmental Genomes
Table S3. Bootstrapped frequency of COG occurrences for specific metabolic processes encoded in the Antarctic bacterioplankton environmental genomes. Category COG Predicted Protein WEG SEG Inorganic carbon COG1850 Ribulose 1,5-bisphosphate carboxylase, large subunit 4.34 0.85 COG4451 Ribulose bisphosphate carboxylase small subunit 1.19 0.83 COG0574 Phosphoenolpyruvate synthase/pyruvate phosphate dikinase 13.7 6.63 COG4770 Acetyl/propionyl-CoA carboxylase, alpha subunit 7.91 4.85 Sulfur Metabolism (inorganic) COG2897 Rhodanese-related sulfurtransferase 5.64 3.25 COG0607 Rhodanese-related sulfurtransferase 6.02 5.68 COG2895 GTPases - Sulfate adenylate transferase subunit 1 2.28 4.88 COG1054 Predicted sulfurtransferase 2.53 4.17 COG0306 Phosphate/sulphate permeases 7.56 3.27 COG0659 Sulfate permease and related transporters (MFS superfamily) 15.72 15.22 COG0529 Adenylylsulfate kinase and related kinases 1.98 4.16 COG0225 Peptide methionine sulfoxide reductase 7.03 3.22 COG0229 Conserved domain frequently associated with peptide methionine sulfoxide reductase 3.77 2.47 COG0641 Arylsulfatase regulator (Fe-S oxidoreductase) 0.67 0 COG0526 Thiol-disulfide isomerase and thioredoxins 6.99 4.12 COG1651 Protein-disulfide isomerase 10.75 1.65 COG2041 Sulfite oxidase and related enzymes 5.51 3.05 COG2046 ATP sulfurylase (sulfate adenylyltransferase) 4.34 3.46 COG2221 Dissimilatory sulfite reductase (desulfoviridin), alpha and beta subunits 1.21 0 COG2920 Dissimilatory sulfite reductase (desulfoviridin), gamma subunit 1.9 0 COG0155 Sulfite reductase, -
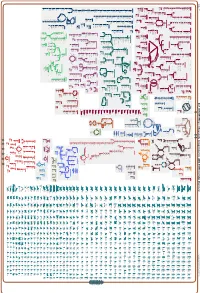
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_003855395Cyc: Shewanella livingstonensis LMG 19866 Cellular Overview Connections between pathways are omitted for legibility. -

Mitochondria Targeting As an Effective Strategy for Cancer Therapy
International Journal of Molecular Sciences Review Mitochondria Targeting as an Effective Strategy for Cancer Therapy Poorva Ghosh , Chantal Vidal, Sanchareeka Dey and Li Zhang * Department of Biological Sciences, the University of Texas at Dallas, Richardson, TX 75080, USA; [email protected] (P.G.); [email protected] (C.V.); [email protected] (S.D.) * Correspondence: [email protected]; Tel.: +972-883-5757 Received: 25 February 2020; Accepted: 6 May 2020; Published: 9 May 2020 Abstract: Mitochondria are well known for their role in ATP production and biosynthesis of macromolecules. Importantly, increasing experimental evidence points to the roles of mitochondrial bioenergetics, dynamics, and signaling in tumorigenesis. Recent studies have shown that many types of cancer cells, including metastatic tumor cells, therapy-resistant tumor cells, and cancer stem cells, are reliant on mitochondrial respiration, and upregulate oxidative phosphorylation (OXPHOS) activity to fuel tumorigenesis. Mitochondrial metabolism is crucial for tumor proliferation, tumor survival, and metastasis. Mitochondrial OXPHOS dependency of cancer has been shown to underlie the development of resistance to chemotherapy and radiotherapy. Furthermore, recent studies have demonstrated that elevated heme synthesis and uptake leads to intensified mitochondrial respiration and ATP generation, thereby promoting tumorigenic functions in non-small cell lung cancer (NSCLC) cells. Also, lowering heme uptake/synthesis inhibits mitochondrial OXPHOS and effectively reduces oxygen consumption, thereby inhibiting cancer cell proliferation, migration, and tumor growth in NSCLC. Besides metabolic changes, mitochondrial dynamics such as fission and fusion are also altered in cancer cells. These alterations render mitochondria a vulnerable target for cancer therapy. This review summarizes recent advances in the understanding of mitochondrial alterations in cancer cells that contribute to tumorigenesis and the development of drug resistance. -

Associated Gene Expression Profiles in Multiple Sclerosis Jeroen Melief1, Marie Orre2, Koen Bossers3, Corbert G
Melief et al. Acta Neuropathologica Communications (2019) 7:60 https://doi.org/10.1186/s40478-019-0705-7 RESEARCH Open Access Transcriptome analysis of normal-appearing white matter reveals cortisol- and disease- associated gene expression profiles in multiple sclerosis Jeroen Melief1, Marie Orre2, Koen Bossers3, Corbert G. van Eden1, Karianne G. Schuurman1, Matthew R. J. Mason1,3, Joost Verhaagen3, Jörg Hamann1,4 and Inge Huitinga1* Abstract Inter-individual differences in cortisol production by the hypothalamus–pituitary–adrenal (HPA) axis are thought to contribute to clinical and pathological heterogeneity of multiple sclerosis (MS). At the same time, accumulating evidence indicates that MS pathogenesis may originate in the normal-appearing white matter (NAWM). Therefore, we performed a genome-wide transcriptional analysis, by Agilent microarray, of post-mortem NAWM of 9 control subjects and 18 MS patients to investigate to what extent gene expression reflects disease heterogeneity and HPA- axis activity. Activity of the HPA axis was determined by cortisol levels in cerebrospinal fluid and by numbers of corticotropin-releasing neurons in the hypothalamus, while duration of MS and time to EDSS6 served as indicator of disease severity. Applying weighted gene co-expression network analysis led to the identification of a range of gene modules with highly similar co-expression patterns that strongly correlated with various indicators of HPA-axis activity and/or severity of MS. Interestingly, molecular profiles associated with relatively