Restriction of Aerobic Metabolism by Acquired Or Innate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Basis of Simple and Complex Traits with Relevance to Avian Evolution
Genetic basis of simple and complex traits with relevance to avian evolution Małgorzata Anna Gazda Doctoral Program in Biodiversity, Genetics and Evolution D Faculdade de Ciências da Universidade do Porto 2019 Supervisor Miguel Jorge Pinto Carneiro, Auxiliary Researcher, CIBIO/InBIO, Laboratório Associado, Universidade do Porto Co-supervisor Ricardo Lopes, CIBIO/InBIO Leif Andersson, Uppsala University FCUP Genetic basis of avian traits Nota Previa Na elaboração desta tese, e nos termos do número 2 do Artigo 4º do Regulamento Geral dos Terceiros Ciclos de Estudos da Universidade do Porto e do Artigo 31º do D.L.74/2006, de 24 de Março, com a nova redação introduzida pelo D.L. 230/2009, de 14 de Setembro, foi efetuado o aproveitamento total de um conjunto coerente de trabalhos de investigação já publicados ou submetidos para publicação em revistas internacionais indexadas e com arbitragem científica, os quais integram alguns dos capítulos da presente tese. Tendo em conta que os referidos trabalhos foram realizados com a colaboração de outros autores, o candidato esclarece que, em todos eles, participou ativamente na sua conceção, na obtenção, análise e discussão de resultados, bem como na elaboração da sua forma publicada. Este trabalho foi apoiado pela Fundação para a Ciência e Tecnologia (FCT) através da atribuição de uma bolsa de doutoramento (PD/BD/114042/2015) no âmbito do programa doutoral em Biodiversidade, Genética e Evolução (BIODIV). 2 FCUP Genetic basis of avian traits Acknowledgements Firstly, I would like to thank to my all supervisors Miguel Carneiro, Ricardo Lopes and Leif Andersson, for the demanding task of supervising myself last four years. -

EGFR Confers Exquisite Specificity of Wnt9a-Fzd9b Signaling in Hematopoietic Stem Cell Development
bioRxiv preprint doi: https://doi.org/10.1101/387043; this version posted August 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Grainger, et al, 2018 EGFR confers exquisite specificity of Wnt9a-Fzd9b signaling in hematopoietic stem cell development Stephanie Grainger1, Nicole Nguyen1, Jenna Richter1,2, Jordan Setayesh1, Brianna Lonquich1, Chet Huan Oon1, Jacob M. Wozniak2,3,4, Rocio Barahona1, Caramai N. Kamei5, Jack Houston1,2, Marvic Carrillo-Terrazas3,4, Iain A. Drummond5,6, David Gonzalez3.4, Karl Willert#,¥,1, and David Traver¥,1,7. ¥co-corresponding authors: [email protected]; [email protected] #Lead contact 1Department of Cellular and Molecular Medicine, University of California, San Diego, La Jolla, California, 92037, USA. 2Biomedical Sciences Graduate Program, University of California, San Diego, La Jolla, California, 92037, USA. 3Skaggs School of Pharmacy and Pharmaceutical Science, University of California, San Diego, La Jolla, California, 92093, USA. 4Department of Pharmacology, University of California, San Diego, La Jolla, California, 92092 5Massachusetts General Hospital Nephrology Division, Charlestown, Massachusetts, 02129, USA. 6Harvard Medical School, Department of Genetics, Boston MA 02115 7Section of Cell and Developmental Biology, University of California, San Diego, La Jolla, California, 92037, USA. Running title: A mechanism for Wnt-Fzd specificity in hematopoietic stem cells Keywords: hematopoietic stem cell (HSC), Wnt, Wnt9a, human, zebrafish, Fzd, Fzd9b, FZD9, EGFR, APEX2 1 bioRxiv preprint doi: https://doi.org/10.1101/387043; this version posted August 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Dual Regulation of Planar Polarization by Secreted Wnts and Vangl2 in the Developing Mouse Cochlea Elvis Huarcaya Najarro1, Jennifer Huang1, Adrian Jacobo2, Lee A
© 2020. Published by The Company of Biologists Ltd | Development (2020) 147, dev191981. doi:10.1242/dev.191981 RESEARCH ARTICLE Dual regulation of planar polarization by secreted Wnts and Vangl2 in the developing mouse cochlea Elvis Huarcaya Najarro1, Jennifer Huang1, Adrian Jacobo2, Lee A. Quiruz1, Nicolas Grillet1 and Alan G. Cheng1,* ABSTRACT on the other. Flamingo (Fmi/Celsr1, Fmi/Celsr2 and Fmi/Celsr3) is Planar cell polarity (PCP) proteins localize asymmetrically to instruct present on both poles of the cell. Defective PCP signaling cell polarity within the tissue plane, with defects leading to deformities represented by a lack of polarized PCP components leads to of the limbs, neural tube and inner ear. Wnt proteins are evolutionarily congenital heart and tracheal abnormalities, skeletal dysplasia, conserved polarity cues, yet Wnt mutants display variable PCP neural tube defects as well as cochlear deformities (Butler and defects; thus, how Wnts regulate PCP remains unresolved. Here, we Wallingford, 2017; White et al., 2018). Despite their crucial roles, have used the developing cochlea as a model system to show that our understanding of upstream signals orchestrating PCP signaling secreted Wnts regulate PCP through polarizing a specific subset of is rather limited. PCP proteins. Conditional deletion of Wntless or porcupine, both of Wnt proteins have been implicated as upstream polarity cues for which are essential for secretion of Wnts, caused misrotated sensory PCP signaling. For example, limb morphogenesis in mice requires a cells and shortened cochlea – both hallmarks of PCP defects. gradient of Wnt5a, which has been reported to act as an instructive Wntless-deficient cochleae lacked the polarized PCP components cue to establish PCP (Gao et al., 2018, 2011). -

A General Binding Mechanism for All Human Sulfatases by the Formylglycine-Generating Enzyme
A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme Dirk Roeser*, Andrea Preusser-Kunze†, Bernhard Schmidt†, Kathrin Gasow*, Julia G. Wittmann*, Thomas Dierks‡, Kurt von Figura†, and Markus Georg Rudolph*§ *Department of Molecular Structural Biology, University of Go¨ttingen, Justus-von-Liebig-Weg 11, D-37077 Go¨ttingen, Germany; †Department of Biochemistry II, Heinrich-Du¨ker-Weg 12, University of Go¨ttingen, D-37073 Go¨ttingen, Germany; and ‡Department of Biochemistry I, Universita¨tsstrasse 25, University of Bielefeld, D-33615 Bielefeld, Germany Edited by Carolyn R. Bertozzi, University of California, Berkeley, CA, and approved November 8, 2005 (received for review September 1, 2005) The formylglycine (FGly)-generating enzyme (FGE) uses molecular tases, suggesting a general binding mechanism of substrate sulfa- oxygen to oxidize a conserved cysteine residue in all eukaryotic tases by FGE. sulfatases to the catalytically active FGly. Sulfatases degrade and The details of how O2-dependent cysteine oxidation is mediated remodel sulfate esters, and inactivity of FGE results in multiple by FGE are unknown. As a first step toward the elucidation of the sulfatase deficiency, a fatal disease. The previously determined FGE molecular mechanism of FGly formation, we have previously crystal structure revealed two crucial cysteine residues in the active determined crystal structures of FGE in various oxidation states site, one of which was thought to be implicated in substrate (8). FGE adopts a novel fold with surprisingly little regular sec- 2ϩ binding. The other cysteine residue partakes in a novel oxygenase ondary structure and contains two structural Ca ions and two mechanism that does not rely on any cofactors. -

WNT Signalling in Prostate Cancer
WNT signalling in prostate cancer Virginia Murillo-Garzón1 and Robert Kypta1,2 1Cell Biology and Stem Cells Unit, CIC bioGUNE, Building 801A, Bizkaia Technology Park, Derio 48160, Spain 2Department of Surgery and Cancer, Imperial College London, Du Cane Road, London W12 0NN, UK Biographies: Robert Kypta is a Principal Investigator at CIC bioGUNE, a Centre of Excellence Severo Ochoa near Bilbao, and a Lecturer in Prostate Cancer at Imperial College London. His research interests focus on hoW extracellular signals control cell fate during prostate cancer progression and neuronal differentiation. Virginia Murillo Garzón is a PhD student at CIC bioGUNE, a Centre of Excellence Severo Ochoa near Bilbao. She has BSc in Biotechnology from the University of Salamanca and a Masters in Regenerative Biomedicine from the University of Granada. Her PhD is on Wnt receptor signalling in prostate cancer. 1 Abstract Genome sequencing and gene expression analyses of prostate tumours have highlighted the potential importance of genetic and epigenetic changes observed in WNT signalling pathWay components in prostate tumours, particularly in the development of castration-resistant prostate cancer. WNT signalling is also important in the prostate tumour microenvironment, Where WNT proteins secreted by the tumour stroma promote therapy resistance, and in prostate cancer stem or progenitor cells, Where WNT-b-catenin signals promote self-reneWal or expansion. Preclinical studies have demonstrated the potential of inhibitors that target WNT-receptor complexes at the membrane or that block the interaction of b-catenin with LEF1 and the androgen receptor, in preventing prostate cancer progression. Some Wnt signalling inhibitors are in Phase I trials, but they have yet to be tested in patients With prostate cancer. -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Lysosomal Hydrolases: Conversion of Acidic to Basic Forms by Neuraminidase
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 13, number 1 FEBS LETTERS February 1971 LYSOSOMAL HYDROLASES: CONVERSION OF ACIDIC TO BASIC FORMS BY NEURAMINIDASE Alfred GOLDSTONE, Philip KONECNY and Harold KOENIG Neurology Service, VA Research Hospital; and Departments of Neurology and Biochemistry, Northwestern University Medical School, Chicago, Illinois, USA Received 21 December 1970 1. Introduction intervals to avoid overheating). The lysosomal lysate was ultracentrifuged at 100,000 g for 30 min to give A number of hydrolytic enzymes of lysosomal a soluble enzyme fraction. This fraction also contains origin occur in multiple forms which differ in such an acidic lipoprotein [4]. The insoluble residue was physicochemical properties as solubility, heat and pH suspended in 0.05 M acetate buffer pH 5.0 with 0.2% stability, pH optima and electrophoretic mobility [I]. Triton X-100 and resonicated to release the bound ly- Recent studies in this laboratory indicate that essen- sosomal enzymes together with additional acidic lipo- tially all the enzyme proteins of rat kidney and liver protein ([4] and A. Goldstone and H. Koenig, unpu- lysosomes are glycoproteins, and that at least some of blished observations). All operations were carried out these glycoproteins contain sialic acid [2-41. The at 4’. acidic form of lysosomal N-acetylhexosaminidase from Aliquots of the soluble fraction containing about human spleen [ 51 and kidney [6] is converted into 2 mg of protein were incubated with neuraminidase the basic form by treatment with neuraminidase. We from Cl. perfringens (Type Vl enzyme supplied by now confirm this finding for rat kidney and show Sigma), 0.1 mg/ml, or V. -

Towards an Integrated View of Wnt Signaling in Development Renée Van Amerongen and Roel Nusse*
HYPOTHESIS 3205 Development 136, 3205-3214 (2009) doi:10.1242/dev.033910 Towards an integrated view of Wnt signaling in development Renée van Amerongen and Roel Nusse* Wnt signaling is crucial for embryonic development in all animal Notably, components at virtually every level of the Wnt signal species studied to date. The interaction between Wnt proteins transduction cascade have been shown to affect both β-catenin- and cell surface receptors can result in a variety of intracellular dependent and -independent responses, depending on the cellular responses. A key remaining question is how these specific context. As we discuss below, this holds true for the Wnt proteins responses take shape in the context of a complex, multicellular themselves, as well as for their receptors and some intracellular organism. Recent studies suggest that we have to revise some of messengers. Rather than concluding that these proteins are shared our most basic ideas about Wnt signal transduction. Rather than between pathways, we instead propose that it is the total net thinking about Wnt signaling in terms of distinct, linear, cellular balance of signals that ultimately determines the response of the signaling pathways, we propose a novel view that considers the receiving cell. In the context of an intact and developing integration of multiple, often simultaneous, inputs at the level organism, cells receive multiple, dynamic, often simultaneous and of both Wnt-receptor binding and the downstream, sometimes even conflicting inputs, all of which are integrated to intracellular response. elicit the appropriate cell behavior in response. As such, the different signaling pathways might thus be more intimately Introduction intertwined than previously envisioned. -

Increased CHST15 Follows Decline in Arylsulfatase B (ARSB) and Disinhibition of Non-Canonical WNT Signaling: Potential Impact on Epithelial and Mesenchymal Identity
www.oncotarget.com Oncotarget, 2020, Vol. 11, (No. 24), pp: 2327-2344 Research Paper Increased CHST15 follows decline in arylsulfatase B (ARSB) and disinhibition of non-canonical WNT signaling: potential impact on epithelial and mesenchymal identity Sumit Bhattacharyya1,2, Leo Feferman1,2, Xiaorui Han3, Ke Xia3, Fuming Zhang3, Robert J. Linhardt3, Joanne K. Tobacman1,2 1Department of Medicine, University of Illinois at Chicago, Chicago, IL, USA 2Jesse Brown VAMC, Chicago, IL, USA 3Department of Chemistry and Chemical Biology Rensselaer Polytechnic Insitute, Troy, NY, USA Correspondence to: Joanne K. Tobacman, email: [email protected] Keywords: sulfotransferase; sulfatase; chondroitin sulfate; Wnt; EMT Received: February 18, 2020 Accepted: May 20, 2020 Published: June 16, 2020 Copyright: Bhattacharyya et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Expression of CHST15 (carbohydrate sulfotransferase 15; chondroitin 4-sulfate- 6-sulfotransferase; BRAG), the sulfotransferase enzyme that adds 6-sulfate to chondroitin 4-sulfate (C4S) to make chondroitin 4,6-disulfate (chondroitin sulfate E, CSE), was increased in malignant prostate epithelium obtained by laser capture microdissection and following arylsulfatase B (ARSB; N-acetylgalactosamine- 4-sulfatase) silencing in human prostate epithelial cells. Experiments in normal and malignant human prostate epithelial and stromal cells and tissues, in HepG2 cells, and in the ARSB-null mouse were performed to determine the pathway by which CHST15 expression is up-regulated when ARSB expression is reduced. Effects of Wnt- containing prostate stromal cell spent media and selective inhibitors of WNT, JNK, p38, SHP2, β-catenin, Rho, and Rac-1 signaling pathways were determined. -

Early Growth Response 1 Regulates Hematopoietic Support and Proliferation in Human Primary Bone Marrow Stromal Cells
Hematopoiesis SUPPLEMENTARY APPENDIX Early growth response 1 regulates hematopoietic support and proliferation in human primary bone marrow stromal cells Hongzhe Li, 1,2 Hooi-Ching Lim, 1,2 Dimitra Zacharaki, 1,2 Xiaojie Xian, 2,3 Keane J.G. Kenswil, 4 Sandro Bräunig, 1,2 Marc H.G.P. Raaijmakers, 4 Niels-Bjarne Woods, 2,3 Jenny Hansson, 1,2 and Stefan Scheding 1,2,5 1Division of Molecular Hematology, Department of Laboratory Medicine, Lund University, Lund, Sweden; 2Lund Stem Cell Center, Depart - ment of Laboratory Medicine, Lund University, Lund, Sweden; 3Division of Molecular Medicine and Gene Therapy, Department of Labora - tory Medicine, Lund University, Lund, Sweden; 4Department of Hematology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands and 5Department of Hematology, Skåne University Hospital Lund, Skåne, Sweden ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2019.216648 Received: January 14, 2019. Accepted: July 19, 2019. Pre-published: August 1, 2019. Correspondence: STEFAN SCHEDING - [email protected] Li et al.: Supplemental data 1. Supplemental Materials and Methods BM-MNC isolation Bone marrow mononuclear cells (BM-MNC) from BM aspiration samples were isolated by density gradient centrifugation (LSM 1077 Lymphocyte, PAA, Pasching, Austria) either with or without prior incubation with RosetteSep Human Mesenchymal Stem Cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada) for lineage depletion (CD3, CD14, CD19, CD38, CD66b, glycophorin A). BM-MNCs from fetal long bones and adult hip bones were isolated as reported previously 1 by gently crushing bones (femora, tibiae, fibulae, humeri, radii and ulna) in PBS+0.5% FCS subsequent passing of the cell suspension through a 40-µm filter. -

PFK-2/Fbpase-2: Maker and Breaker of the Essential Biofactor Fructose-2, 6-Bisphosphate
30 Review TRENDS in Biochemical Sciences Vol.26 No.1 January 2001 PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2, 6-bisphosphate DavidA. Okar, Ànna Manzano,Aurèa Navarro-Sabatè, Lluìs Riera, Ramon Bartrons and Alex J. Lange Fructose-2,6-bisphosphate is responsible for mediating glucagon-stimulated extraction in base led to the discovery of F-2,6-P2 and gluconeogenesis in the liver.This discovery has led to the realization that this the realization that it was either formed or destroyed compound plays a significant role in directing carbohydrate fluxes in all during metabolic transitions where its concentration eukaryotes. Biophysical studies of the enzyme that both synthesizes and determined changes in glycolytic and gluconeogenic degrades this biofactor have yielded insight into its molecular enzymology. carbon flux in the liver. Interest in this compound grew Moreover, the metabolic role of fructose-2,6-bisphosphate has great potential rapidly because it was found to be a most potent in the treatment of diabetes. positive allosteric effector of 6-phosphofructo-1- kinase (PFK-1; EC 2.7.1.11) and an inhibitor of A trail that began with the discovery of a highly potent fructose-1,6-bisphosphatase (FBPase-1; EC 3.1.3.11). regulator of mammalian hepatic carbohydrate Because of its antagonistic actions on these enzymes, metabolism, fructose-2,6-bisphosphate (F-2,6-P2), led F-2,6-P2 plays a crucial role in the control of the to the discovery of the bifunctional enzyme opposing hepatic glycolytic and gluconeogenic 6-phosphofructo-2-kinase/fructose- pathways1–4 (Fig. -
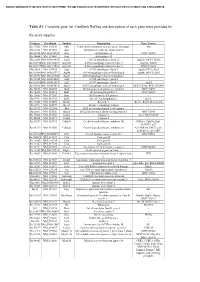
Table S1. Complete Gene List. Genbank Refseq and Description of Each Gene Were Provided By
Document downloaded from http://www.elsevier.es, day 24/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Table S1. Complete gene list. GenBank RefSeq and description of each gene were provided by the array supplier. Unigene GeneBank Symbol Description Gene Name/s Rn.11422 NM_033230 Akt1 V-akt murine thymoma viral oncogene homolog 1 Akt Rn.2104 NM_019288 App Amyloid beta (A4) precursor protein - Rn.23323 NM_001034933 Arsa Arylsulfatase A MGC125207 Rn.94004 NM_033443 Arsb Arylsulfatase B - Rn.6224 NM_001038495 Atg12 ATG12 autophagy related 12 Apg12l, MGC125080 Rn.101734NM_001108809 Atg16l1 ATG16 autophagy related 16-like 1 Apg16l, Wdr30 Rn.104199NM_001191560 Atg16l2 ATG16 autophagy related 16-like 2 RGD1311400 Rn.3084 NM_134394 Atg3 ATG3 autophagy related 3 Apg3l, PIG-1, Pig1 Rn.163086NM_001025711 Atg4b ATG4 autophagy related 4 homolog B Apg4b, MGC112887 Rn.23378 NM_001107948 Atg4c ATG4 autophagy related 4 homolog C - Rn.98385 NM_001014250 Atg5 ATG5 autophagy related 5 - Rn.162765NM_001012097 Atg7 ATG7 autophagy related 7 Apg7l Rn.35248 NM_001014218 Atg9a ATG9 autophagy related 9 homolog A MGC105908, RGD1310450 Rn.36696 NM_022698 Bad BCL2-associated agonist of cell death MGC72439 Rn.14598 NM_053812 Bak1 BCL2-antagonist/killer 1 MGC108627 Rn.10668 NM_017059 Bax Bcl2-associated X protein - Rn.9996 NM_016993 Bcl2 B-cell CLL/lymphoma 2 Bcl-2 Rn.10323 NM_031535 Bcl2l1 Bcl2-like 1 Bcl-xl, Bcl2l, Bclx, bcl-X Rn.2776 NM_053739 Becn1 Beclin 1, autophagy related - Rn.31142 NM_022684