Recombinant Human Arylsulfatase B/ARSB
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Basis of Simple and Complex Traits with Relevance to Avian Evolution
Genetic basis of simple and complex traits with relevance to avian evolution Małgorzata Anna Gazda Doctoral Program in Biodiversity, Genetics and Evolution D Faculdade de Ciências da Universidade do Porto 2019 Supervisor Miguel Jorge Pinto Carneiro, Auxiliary Researcher, CIBIO/InBIO, Laboratório Associado, Universidade do Porto Co-supervisor Ricardo Lopes, CIBIO/InBIO Leif Andersson, Uppsala University FCUP Genetic basis of avian traits Nota Previa Na elaboração desta tese, e nos termos do número 2 do Artigo 4º do Regulamento Geral dos Terceiros Ciclos de Estudos da Universidade do Porto e do Artigo 31º do D.L.74/2006, de 24 de Março, com a nova redação introduzida pelo D.L. 230/2009, de 14 de Setembro, foi efetuado o aproveitamento total de um conjunto coerente de trabalhos de investigação já publicados ou submetidos para publicação em revistas internacionais indexadas e com arbitragem científica, os quais integram alguns dos capítulos da presente tese. Tendo em conta que os referidos trabalhos foram realizados com a colaboração de outros autores, o candidato esclarece que, em todos eles, participou ativamente na sua conceção, na obtenção, análise e discussão de resultados, bem como na elaboração da sua forma publicada. Este trabalho foi apoiado pela Fundação para a Ciência e Tecnologia (FCT) através da atribuição de uma bolsa de doutoramento (PD/BD/114042/2015) no âmbito do programa doutoral em Biodiversidade, Genética e Evolução (BIODIV). 2 FCUP Genetic basis of avian traits Acknowledgements Firstly, I would like to thank to my all supervisors Miguel Carneiro, Ricardo Lopes and Leif Andersson, for the demanding task of supervising myself last four years. -

Tepzz¥5Z5 8 a T
(19) TZZ¥Z___T (11) EP 3 505 181 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 03.07.2019 Bulletin 2019/27 A61K 38/46 (2006.01) C12N 9/16 (2006.01) (21) Application number: 18248241.4 (22) Date of filing: 28.12.2018 (84) Designated Contracting States: (72) Inventors: AL AT BE BG CH CY CZ DE DK EE ES FI FR GB • DICKSON, Patricia GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO Torrance, CA California 90502 (US) PL PT RO RS SE SI SK SM TR • CHOU, Tsui-Fen Designated Extension States: Torrance, CA California 90502 (US) BA ME • EKINS, Sean Designated Validation States: Brooklyn, NY New York 11215 (US) KH MA MD TN • KAN, Shih-Hsin Torrance, CA California 90502 (US) (30) Priority: 28.12.2017 US 201762611472 P • LE, Steven 05.04.2018 US 201815946505 Torrance, CA California 90502 (US) • MOEN, Derek R. (71) Applicants: Torrance, CA California 90502 (US) • Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center (74) Representative: J A Kemp Torrance, CA 90502 (US) 14 South Square • Phoenix Nest Inc. Gray’s Inn Brooklyn NY 11215 (US) London WC1R 5JJ (GB) (54) PREPARATION OF ENZYME REPLACEMENT THERAPY FOR MUCOPOLYSACCHARIDOSIS IIID (57) The present disclosure relates to compositions for use in a method of treating Sanfilippo syndrome (also known as Sanfilippo disease type D, Sanfilippo D, mu- copolysaccharidosis type IIID, MPS IIID). The method can entail injecting to the spinal fluid of a MPS IIID patient an effective amount of a composition comprising a re- combinant human acetylglucosamine-6-sulfatase (GNS) protein comprising the amino acid sequence of SEQ ID NO: 1 or an amino acid sequence having at least 90% sequence identity to SEQ ID NO: 1 and having the en- zymatic activity of the human GNS protein. -

A General Binding Mechanism for All Human Sulfatases by the Formylglycine-Generating Enzyme
A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme Dirk Roeser*, Andrea Preusser-Kunze†, Bernhard Schmidt†, Kathrin Gasow*, Julia G. Wittmann*, Thomas Dierks‡, Kurt von Figura†, and Markus Georg Rudolph*§ *Department of Molecular Structural Biology, University of Go¨ttingen, Justus-von-Liebig-Weg 11, D-37077 Go¨ttingen, Germany; †Department of Biochemistry II, Heinrich-Du¨ker-Weg 12, University of Go¨ttingen, D-37073 Go¨ttingen, Germany; and ‡Department of Biochemistry I, Universita¨tsstrasse 25, University of Bielefeld, D-33615 Bielefeld, Germany Edited by Carolyn R. Bertozzi, University of California, Berkeley, CA, and approved November 8, 2005 (received for review September 1, 2005) The formylglycine (FGly)-generating enzyme (FGE) uses molecular tases, suggesting a general binding mechanism of substrate sulfa- oxygen to oxidize a conserved cysteine residue in all eukaryotic tases by FGE. sulfatases to the catalytically active FGly. Sulfatases degrade and The details of how O2-dependent cysteine oxidation is mediated remodel sulfate esters, and inactivity of FGE results in multiple by FGE are unknown. As a first step toward the elucidation of the sulfatase deficiency, a fatal disease. The previously determined FGE molecular mechanism of FGly formation, we have previously crystal structure revealed two crucial cysteine residues in the active determined crystal structures of FGE in various oxidation states site, one of which was thought to be implicated in substrate (8). FGE adopts a novel fold with surprisingly little regular sec- 2ϩ binding. The other cysteine residue partakes in a novel oxygenase ondary structure and contains two structural Ca ions and two mechanism that does not rely on any cofactors. -

Comparative Study of Idursulfase Beta and Idursulfase in Vitro and in Vivo
Journal of Human Genetics (2017) 62, 167–174 OPEN Official journal of the Japan Society of Human Genetics www.nature.com/jhg ORIGINAL ARTICLE Comparative study of idursulfase beta and idursulfase in vitro and in vivo Chihwa Kim1, Jinwook Seo2, Yokyung Chung2, Hyi-Jeong Ji3, Jaehyeon Lee2, Jongmun Sohn2, Byoungju Lee2 and Eui-cheol Jo1 Hunter syndrome is an X-linked lysosomal storage disease caused by a deficiency in the enzyme iduronate-2-sulfatase (IDS), leading to the accumulation of glycosaminoglycans (GAGs). Two recombinant enzymes, idursulfase and idursulfase beta are currently available for enzyme replacement therapy for Hunter syndrome. These two enzymes exhibited some differences in various clinical parameters in a recent clinical trial. Regarding the similarities and differences of these enzymes, previous research has characterized their biochemical and physicochemical properties. We compared the in vitro and in vivo efficacy of the two enzymes on patient fibroblasts and mouse model. Two enzymes were taken up into the cell and degraded GAGs accumulated in fibroblasts. In vivo studies of two enzymes revealed similar organ distribution and decreased urinary GAGs excretion. Especially, idursulfase beta exhibited enhanced in vitro efficacy for the lower concentration of treatment, in vivo efficacy in the degradation of tissue GAGs and improvement of bones, and revealed lower anti-drug antibody formation. A biochemical analysis showed that both enzymes show largely a similar glycosylation pattern, but the several peaks were different and quantity of aggregates of idursulfase beta was lower. Journal of Human Genetics (2017) 62, 167–174; doi:10.1038/jhg.2016.133; published online 10 November 2016 INTRODUCTION secreted protein and contains eight N-linked glycosylation sites at Mucopolysaccharidosis II (MPS II, Hunter syndrome; OMIM 309900) positions 31, 115, 144, 246, 280, 325, 513 and 537. -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Lysosomal Hydrolases: Conversion of Acidic to Basic Forms by Neuraminidase
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 13, number 1 FEBS LETTERS February 1971 LYSOSOMAL HYDROLASES: CONVERSION OF ACIDIC TO BASIC FORMS BY NEURAMINIDASE Alfred GOLDSTONE, Philip KONECNY and Harold KOENIG Neurology Service, VA Research Hospital; and Departments of Neurology and Biochemistry, Northwestern University Medical School, Chicago, Illinois, USA Received 21 December 1970 1. Introduction intervals to avoid overheating). The lysosomal lysate was ultracentrifuged at 100,000 g for 30 min to give A number of hydrolytic enzymes of lysosomal a soluble enzyme fraction. This fraction also contains origin occur in multiple forms which differ in such an acidic lipoprotein [4]. The insoluble residue was physicochemical properties as solubility, heat and pH suspended in 0.05 M acetate buffer pH 5.0 with 0.2% stability, pH optima and electrophoretic mobility [I]. Triton X-100 and resonicated to release the bound ly- Recent studies in this laboratory indicate that essen- sosomal enzymes together with additional acidic lipo- tially all the enzyme proteins of rat kidney and liver protein ([4] and A. Goldstone and H. Koenig, unpu- lysosomes are glycoproteins, and that at least some of blished observations). All operations were carried out these glycoproteins contain sialic acid [2-41. The at 4’. acidic form of lysosomal N-acetylhexosaminidase from Aliquots of the soluble fraction containing about human spleen [ 51 and kidney [6] is converted into 2 mg of protein were incubated with neuraminidase the basic form by treatment with neuraminidase. We from Cl. perfringens (Type Vl enzyme supplied by now confirm this finding for rat kidney and show Sigma), 0.1 mg/ml, or V. -

Sanfilippo Disease Type D: Deficiency of N-Acetylglucosamine-6- Sulfate Sulfatase Required for Heparan Sulfate Degradation
Proc. Nat!. Acad. Sci. USA Vol. 77, No. 11, pp. 6822-6826, November 1980 Medical Sciences Sanfilippo disease type D: Deficiency of N-acetylglucosamine-6- sulfate sulfatase required for heparan sulfate degradation (mucopolysaccharidosis/keratan sulfate/lysosomes) HANS KRESSE, EDUARD PASCHKE, KURT VON FIGURA, WALTER GILBERG, AND WALBURGA FUCHS Institute of Physiological Chemistry, Waldeyerstrasse 15, D-4400 Mfinster, Federal Republic of Germany Communicated by Elizabeth F. Neufeld, July 10, 1980 ABSTRACT Skin fibroblasts from two patients who had lippo syndromes and excreted excessive amounts of heparan symptoms of the Sanfilippo syndrome (mucopolysaccharidosis sulfate and keratan sulfate in the urine. His fibroblasts were III) accumulated excessive amounts of hean sulfate and were unable to desulfate N-acetylglucosamine-6-sulfate and the unable to release sulfate from N-acety lucosamine)--sulfate linkages in heparan sulfate-derived oligosaccharides. Keratan corresponding sugar alcohol (17, 18). It was therefore suggested sulfate-derived oligosaccharides bearing the same residue at that N-acetylglucosamine-6-sulfate sulfatase is involved in the the nonreducing end and p-nitrophenyl6sulfo-2-acetamido- catabolism of both types of macromolecules. 2-deoxy-D-ucopyranoside were degraded normally. Kinetic In this paper, we describe a new disease, tentatively desig- differences between the sulfatase activities of normal fibro- nated Sanfilippo disease type D, that is characterized by the blasts were found. These observations suggest that N-acetyl- excessive excretion glucosamine4-6sulfate sulfatase activities degrading heparan clinical features of the Sanfilippo syndrome, sulfate and keratan sulfate, respectively, can be distinguished. of heparan sulfate, and the inability to release inorganic sulfate It is the activity directed toward heparan sulfate that is deficient from N-acetylglucosamine-6-sulfate residues of heparan sul- in these patients; we propose that this deficiency causes Sanfi- fate-derived oligosaccharides. -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

Increased CHST15 Follows Decline in Arylsulfatase B (ARSB) and Disinhibition of Non-Canonical WNT Signaling: Potential Impact on Epithelial and Mesenchymal Identity
www.oncotarget.com Oncotarget, 2020, Vol. 11, (No. 24), pp: 2327-2344 Research Paper Increased CHST15 follows decline in arylsulfatase B (ARSB) and disinhibition of non-canonical WNT signaling: potential impact on epithelial and mesenchymal identity Sumit Bhattacharyya1,2, Leo Feferman1,2, Xiaorui Han3, Ke Xia3, Fuming Zhang3, Robert J. Linhardt3, Joanne K. Tobacman1,2 1Department of Medicine, University of Illinois at Chicago, Chicago, IL, USA 2Jesse Brown VAMC, Chicago, IL, USA 3Department of Chemistry and Chemical Biology Rensselaer Polytechnic Insitute, Troy, NY, USA Correspondence to: Joanne K. Tobacman, email: [email protected] Keywords: sulfotransferase; sulfatase; chondroitin sulfate; Wnt; EMT Received: February 18, 2020 Accepted: May 20, 2020 Published: June 16, 2020 Copyright: Bhattacharyya et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Expression of CHST15 (carbohydrate sulfotransferase 15; chondroitin 4-sulfate- 6-sulfotransferase; BRAG), the sulfotransferase enzyme that adds 6-sulfate to chondroitin 4-sulfate (C4S) to make chondroitin 4,6-disulfate (chondroitin sulfate E, CSE), was increased in malignant prostate epithelium obtained by laser capture microdissection and following arylsulfatase B (ARSB; N-acetylgalactosamine- 4-sulfatase) silencing in human prostate epithelial cells. Experiments in normal and malignant human prostate epithelial and stromal cells and tissues, in HepG2 cells, and in the ARSB-null mouse were performed to determine the pathway by which CHST15 expression is up-regulated when ARSB expression is reduced. Effects of Wnt- containing prostate stromal cell spent media and selective inhibitors of WNT, JNK, p38, SHP2, β-catenin, Rho, and Rac-1 signaling pathways were determined. -

Early Growth Response 1 Regulates Hematopoietic Support and Proliferation in Human Primary Bone Marrow Stromal Cells
Hematopoiesis SUPPLEMENTARY APPENDIX Early growth response 1 regulates hematopoietic support and proliferation in human primary bone marrow stromal cells Hongzhe Li, 1,2 Hooi-Ching Lim, 1,2 Dimitra Zacharaki, 1,2 Xiaojie Xian, 2,3 Keane J.G. Kenswil, 4 Sandro Bräunig, 1,2 Marc H.G.P. Raaijmakers, 4 Niels-Bjarne Woods, 2,3 Jenny Hansson, 1,2 and Stefan Scheding 1,2,5 1Division of Molecular Hematology, Department of Laboratory Medicine, Lund University, Lund, Sweden; 2Lund Stem Cell Center, Depart - ment of Laboratory Medicine, Lund University, Lund, Sweden; 3Division of Molecular Medicine and Gene Therapy, Department of Labora - tory Medicine, Lund University, Lund, Sweden; 4Department of Hematology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands and 5Department of Hematology, Skåne University Hospital Lund, Skåne, Sweden ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2019.216648 Received: January 14, 2019. Accepted: July 19, 2019. Pre-published: August 1, 2019. Correspondence: STEFAN SCHEDING - [email protected] Li et al.: Supplemental data 1. Supplemental Materials and Methods BM-MNC isolation Bone marrow mononuclear cells (BM-MNC) from BM aspiration samples were isolated by density gradient centrifugation (LSM 1077 Lymphocyte, PAA, Pasching, Austria) either with or without prior incubation with RosetteSep Human Mesenchymal Stem Cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada) for lineage depletion (CD3, CD14, CD19, CD38, CD66b, glycophorin A). BM-MNCs from fetal long bones and adult hip bones were isolated as reported previously 1 by gently crushing bones (femora, tibiae, fibulae, humeri, radii and ulna) in PBS+0.5% FCS subsequent passing of the cell suspension through a 40-µm filter. -
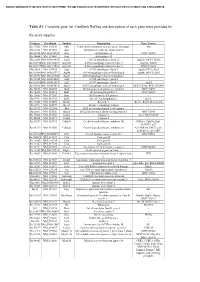
Table S1. Complete Gene List. Genbank Refseq and Description of Each Gene Were Provided By
Document downloaded from http://www.elsevier.es, day 24/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Table S1. Complete gene list. GenBank RefSeq and description of each gene were provided by the array supplier. Unigene GeneBank Symbol Description Gene Name/s Rn.11422 NM_033230 Akt1 V-akt murine thymoma viral oncogene homolog 1 Akt Rn.2104 NM_019288 App Amyloid beta (A4) precursor protein - Rn.23323 NM_001034933 Arsa Arylsulfatase A MGC125207 Rn.94004 NM_033443 Arsb Arylsulfatase B - Rn.6224 NM_001038495 Atg12 ATG12 autophagy related 12 Apg12l, MGC125080 Rn.101734NM_001108809 Atg16l1 ATG16 autophagy related 16-like 1 Apg16l, Wdr30 Rn.104199NM_001191560 Atg16l2 ATG16 autophagy related 16-like 2 RGD1311400 Rn.3084 NM_134394 Atg3 ATG3 autophagy related 3 Apg3l, PIG-1, Pig1 Rn.163086NM_001025711 Atg4b ATG4 autophagy related 4 homolog B Apg4b, MGC112887 Rn.23378 NM_001107948 Atg4c ATG4 autophagy related 4 homolog C - Rn.98385 NM_001014250 Atg5 ATG5 autophagy related 5 - Rn.162765NM_001012097 Atg7 ATG7 autophagy related 7 Apg7l Rn.35248 NM_001014218 Atg9a ATG9 autophagy related 9 homolog A MGC105908, RGD1310450 Rn.36696 NM_022698 Bad BCL2-associated agonist of cell death MGC72439 Rn.14598 NM_053812 Bak1 BCL2-antagonist/killer 1 MGC108627 Rn.10668 NM_017059 Bax Bcl2-associated X protein - Rn.9996 NM_016993 Bcl2 B-cell CLL/lymphoma 2 Bcl-2 Rn.10323 NM_031535 Bcl2l1 Bcl2-like 1 Bcl-xl, Bcl2l, Bclx, bcl-X Rn.2776 NM_053739 Becn1 Beclin 1, autophagy related - Rn.31142 NM_022684 -

A Possible Role for Arylsulfatase G in Dermatan Sulfate Metabolism
International Journal of Molecular Sciences Article A Possible Role for Arylsulfatase G in Dermatan Sulfate Metabolism Aleksandra Poterala-Hejmo 1,*, Adam Golda 2 , Marcin Pacholczyk 1 , Sebastian Student 1, Anna Tylki-Szyma´nska 3 and Anna Lalik 1,* 1 Department of Systems Biology and Engineering, Silesian University of Technology, 44-100 Gliwice, Poland; [email protected] (M.P.); [email protected] (S.S.) 2 Department of Cardiology, 4th Municipal Hospital, 44-100 Gliwice, Poland; [email protected] 3 Department of Pediatrics, Nutrition and Metabolic Diseases, The Children’s Memorial Health Institute, 04-730 Warsaw, Poland; [email protected] * Correspondence: [email protected] (A.P.-H.); [email protected] (A.L.); Tel.: +48-32-2371168 (A.P.-H.); +48-32-2372769 (A.L.) Received: 2 June 2020; Accepted: 6 July 2020; Published: 12 July 2020 Abstract: Perturbations of glycosaminoglycan metabolism lead to mucopolysaccharidoses (MPS)—lysosomal storage diseases. One type of MPS (type VI) is associated with a deficiency of arylsulfatase B (ARSB), for which we previously established a cellular model using pulmonary artery endothelial cells with a silenced ARSB gene. Here, we explored the effects of silencing the ARSB gene on the growth of human pulmonary artery smooth muscle cells in the presence of different concentrations of dermatan sulfate (DS). The viability of pulmonary artery smooth muscle cells with a silenced ARSB gene was stimulated by the dermatan sulfate. In contrast, the growth of pulmonary artery endothelial cells was not affected. As shown by microarray analysis, the expression of the arylsulfatase G (ARSG) in pulmonary artery smooth muscle cells increased after silencing the arylsulfatase B gene, but the expression of genes encoding other enzymes involved in the degradation of dermatan sulfate did not.