WNT Signalling in Prostate Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

EGFR Confers Exquisite Specificity of Wnt9a-Fzd9b Signaling in Hematopoietic Stem Cell Development
bioRxiv preprint doi: https://doi.org/10.1101/387043; this version posted August 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Grainger, et al, 2018 EGFR confers exquisite specificity of Wnt9a-Fzd9b signaling in hematopoietic stem cell development Stephanie Grainger1, Nicole Nguyen1, Jenna Richter1,2, Jordan Setayesh1, Brianna Lonquich1, Chet Huan Oon1, Jacob M. Wozniak2,3,4, Rocio Barahona1, Caramai N. Kamei5, Jack Houston1,2, Marvic Carrillo-Terrazas3,4, Iain A. Drummond5,6, David Gonzalez3.4, Karl Willert#,¥,1, and David Traver¥,1,7. ¥co-corresponding authors: [email protected]; [email protected] #Lead contact 1Department of Cellular and Molecular Medicine, University of California, San Diego, La Jolla, California, 92037, USA. 2Biomedical Sciences Graduate Program, University of California, San Diego, La Jolla, California, 92037, USA. 3Skaggs School of Pharmacy and Pharmaceutical Science, University of California, San Diego, La Jolla, California, 92093, USA. 4Department of Pharmacology, University of California, San Diego, La Jolla, California, 92092 5Massachusetts General Hospital Nephrology Division, Charlestown, Massachusetts, 02129, USA. 6Harvard Medical School, Department of Genetics, Boston MA 02115 7Section of Cell and Developmental Biology, University of California, San Diego, La Jolla, California, 92037, USA. Running title: A mechanism for Wnt-Fzd specificity in hematopoietic stem cells Keywords: hematopoietic stem cell (HSC), Wnt, Wnt9a, human, zebrafish, Fzd, Fzd9b, FZD9, EGFR, APEX2 1 bioRxiv preprint doi: https://doi.org/10.1101/387043; this version posted August 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Dual Regulation of Planar Polarization by Secreted Wnts and Vangl2 in the Developing Mouse Cochlea Elvis Huarcaya Najarro1, Jennifer Huang1, Adrian Jacobo2, Lee A
© 2020. Published by The Company of Biologists Ltd | Development (2020) 147, dev191981. doi:10.1242/dev.191981 RESEARCH ARTICLE Dual regulation of planar polarization by secreted Wnts and Vangl2 in the developing mouse cochlea Elvis Huarcaya Najarro1, Jennifer Huang1, Adrian Jacobo2, Lee A. Quiruz1, Nicolas Grillet1 and Alan G. Cheng1,* ABSTRACT on the other. Flamingo (Fmi/Celsr1, Fmi/Celsr2 and Fmi/Celsr3) is Planar cell polarity (PCP) proteins localize asymmetrically to instruct present on both poles of the cell. Defective PCP signaling cell polarity within the tissue plane, with defects leading to deformities represented by a lack of polarized PCP components leads to of the limbs, neural tube and inner ear. Wnt proteins are evolutionarily congenital heart and tracheal abnormalities, skeletal dysplasia, conserved polarity cues, yet Wnt mutants display variable PCP neural tube defects as well as cochlear deformities (Butler and defects; thus, how Wnts regulate PCP remains unresolved. Here, we Wallingford, 2017; White et al., 2018). Despite their crucial roles, have used the developing cochlea as a model system to show that our understanding of upstream signals orchestrating PCP signaling secreted Wnts regulate PCP through polarizing a specific subset of is rather limited. PCP proteins. Conditional deletion of Wntless or porcupine, both of Wnt proteins have been implicated as upstream polarity cues for which are essential for secretion of Wnts, caused misrotated sensory PCP signaling. For example, limb morphogenesis in mice requires a cells and shortened cochlea – both hallmarks of PCP defects. gradient of Wnt5a, which has been reported to act as an instructive Wntless-deficient cochleae lacked the polarized PCP components cue to establish PCP (Gao et al., 2018, 2011). -

Towards an Integrated View of Wnt Signaling in Development Renée Van Amerongen and Roel Nusse*
HYPOTHESIS 3205 Development 136, 3205-3214 (2009) doi:10.1242/dev.033910 Towards an integrated view of Wnt signaling in development Renée van Amerongen and Roel Nusse* Wnt signaling is crucial for embryonic development in all animal Notably, components at virtually every level of the Wnt signal species studied to date. The interaction between Wnt proteins transduction cascade have been shown to affect both β-catenin- and cell surface receptors can result in a variety of intracellular dependent and -independent responses, depending on the cellular responses. A key remaining question is how these specific context. As we discuss below, this holds true for the Wnt proteins responses take shape in the context of a complex, multicellular themselves, as well as for their receptors and some intracellular organism. Recent studies suggest that we have to revise some of messengers. Rather than concluding that these proteins are shared our most basic ideas about Wnt signal transduction. Rather than between pathways, we instead propose that it is the total net thinking about Wnt signaling in terms of distinct, linear, cellular balance of signals that ultimately determines the response of the signaling pathways, we propose a novel view that considers the receiving cell. In the context of an intact and developing integration of multiple, often simultaneous, inputs at the level organism, cells receive multiple, dynamic, often simultaneous and of both Wnt-receptor binding and the downstream, sometimes even conflicting inputs, all of which are integrated to intracellular response. elicit the appropriate cell behavior in response. As such, the different signaling pathways might thus be more intimately Introduction intertwined than previously envisioned. -

Increased CHST15 Follows Decline in Arylsulfatase B (ARSB) and Disinhibition of Non-Canonical WNT Signaling: Potential Impact on Epithelial and Mesenchymal Identity
www.oncotarget.com Oncotarget, 2020, Vol. 11, (No. 24), pp: 2327-2344 Research Paper Increased CHST15 follows decline in arylsulfatase B (ARSB) and disinhibition of non-canonical WNT signaling: potential impact on epithelial and mesenchymal identity Sumit Bhattacharyya1,2, Leo Feferman1,2, Xiaorui Han3, Ke Xia3, Fuming Zhang3, Robert J. Linhardt3, Joanne K. Tobacman1,2 1Department of Medicine, University of Illinois at Chicago, Chicago, IL, USA 2Jesse Brown VAMC, Chicago, IL, USA 3Department of Chemistry and Chemical Biology Rensselaer Polytechnic Insitute, Troy, NY, USA Correspondence to: Joanne K. Tobacman, email: [email protected] Keywords: sulfotransferase; sulfatase; chondroitin sulfate; Wnt; EMT Received: February 18, 2020 Accepted: May 20, 2020 Published: June 16, 2020 Copyright: Bhattacharyya et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Expression of CHST15 (carbohydrate sulfotransferase 15; chondroitin 4-sulfate- 6-sulfotransferase; BRAG), the sulfotransferase enzyme that adds 6-sulfate to chondroitin 4-sulfate (C4S) to make chondroitin 4,6-disulfate (chondroitin sulfate E, CSE), was increased in malignant prostate epithelium obtained by laser capture microdissection and following arylsulfatase B (ARSB; N-acetylgalactosamine- 4-sulfatase) silencing in human prostate epithelial cells. Experiments in normal and malignant human prostate epithelial and stromal cells and tissues, in HepG2 cells, and in the ARSB-null mouse were performed to determine the pathway by which CHST15 expression is up-regulated when ARSB expression is reduced. Effects of Wnt- containing prostate stromal cell spent media and selective inhibitors of WNT, JNK, p38, SHP2, β-catenin, Rho, and Rac-1 signaling pathways were determined. -

Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells
BASIC RESEARCH www.jasn.org Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells Congwei Luo,1 Shan Zhou,1 Zhanmei Zhou,1 Yahong Liu,1 Li Yang,1 Jiafeng Liu,1 Yunfang Zhang,2 Hongyan Li,2 Youhua Liu ,1,3 Fan Fan Hou,1 and Lili Zhou1 1State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Division of Nephrology, Nanfang Hospital and 2Department of Nephrology, Huadu District People’s Hospital, Southern Medical University, Guangzhou, China; and 3Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania ABSTRACT Cellular senescence is associated with renal disease progression, and accelerated tubular cell senescence promotes the pathogenesis of renal fibrosis. However, the underlying mechanism is unknown. We assessed the potential role of Wnt9a in tubular cell senescence and renal fibrosis. Compared with tubular cells of normal subjects, tubular cells of humans with a variety of nephropathies and those of several mouse models of CKD expressed high levels of Wnt9a that colocalized with the senescence-related protein p16INK4A. Wnt9a expression level correlated with the extent of renal fibrosis, decline of eGFR, and ex- pression of p16INK4A. Furthermore, ectopic expression of Wnt9a after ischemia-reperfusion injury (IRI) induced activation of b-catenin and exacerbated renal fibrosis. Overexpression of Wnt9a exacerbated tubular senescence, evidenced by increased detection of p16INK4A expression and senescence-associated b-galactosidase activity. Conversely, shRNA-mediated knockdown of Wnt9a repressed IRI-induced renal fibrosis in vivo and impeded the growth of senescent tubular epithelial cells in culture. Notably, Wnt9a- induced renal fibrosis was inhibited by shRNA-mediated silencing of p16INK4A in the IRI mouse model. -
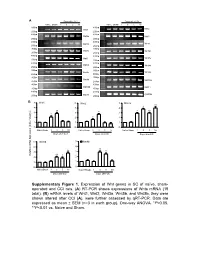
Supplementary Figure 1. Expression of Wnt Genes in SC of Naïve
A Days after CCI Days after CCI Naive Sham 1 3 5 10 Naive Sham 1 3 5 10 500bp 400bp Wnt1 Wnt2 250bp 250bp 400bp 400bp Wnt2b Wnt3 250bp 250bp 400bp 400bp Wnt3a Wnt4 250bp 250bp 300bp 400bp Wnt5a Wnt5b 200bp 250bp 400bp 400bp Wnt6 Wnt7a 250bp 250bp 400bp 400bp Wnt7b Wnt8a 250bp 250bp 400bp 350bp Wnt8b Wnt9a 250bp 200bp 400bp 400bp Wnt9b Wnt10a 250bp 250bp 400bp 400bp Wnt10b Wnt11 250bp 250bp 400bp 400bp Wnt16 GAPDH 250bp 250bp B 5 Wnt1 5 Wnt2 5 Wnt3a ** 4 4 4 * ** ** 3 3 * 3 * * 2 2 * 2 1 1 1 0 0 0 NaïveSham 1 3 5 10 Naïve Sham 1 3 5 10 Naïve Sham 1 3 5 10 Days after CCI Days after CCI Days after CCI 5 Wnt5b 5 Wnt8b 4 4 ** 3 ** 3 ** Relative mRNA Expression (fold of change) ** 2 * 2 1 1 0 0 NaïveSham 1 3 5 10 NaïveSham 1 3 5 10 Days after CCI Days after CCI Supplementary Figure 1 A B Negative Weak Moderate Strong Negative Weak Moderate Strong Large-sized cells 100% 100% 80% 80% 60% 60% 40% 20% 40% 0% 20% Sham CCI-1d CCI-14d Medium-sized cells Wnt3a immunoreactivity cells 0% 100% CGRP(+) IB4(+) 80% Small cells 60% 40% 20% 0% Sham CCI-1d CCI-14d Wnt3a immunoreactivity cells Small cells 100% 80% 60% 40% 20% 0% Sham CCI-1d CCI-14d Fig. S2 4 Fz1 4 Fz3 4 Fz4 4 Fz5 ** 3 3 ** 3 3 2 ** 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1 5 10 NaïveSham 1 5 10 Days after CCI Days after CCI Days after CCI Days after CCI 4 Fz6 4 Fz7 4 Fz8 4 Fz9 (Fold of Change) of (Fold 3 3 3 ** 3 Relative mRNA Expression Expression mRNA Relative ** 2 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1d 5d 10d NaïveSham 1 -

Genome-Wide Screen Reveals WNT11, a Non-Canonical WNT Gene, As a Direct Target of ETS Transcription Factor ERG
Oncogene (2011) 30, 2044–2056 & 2011 Macmillan Publishers Limited All rights reserved 0950-9232/11 www.nature.com/onc ORIGINAL ARTICLE Genome-wide screen reveals WNT11, a non-canonical WNT gene, as a direct target of ETS transcription factor ERG LH Mochmann1, J Bock1, J Ortiz-Ta´nchez1, C Schlee1, A Bohne1, K Neumann2, WK Hofmann3, E Thiel1 and CD Baldus1 1Department of Hematology and Oncology, Charite´, Campus Benjamin Franklin, Berlin, Germany; 2Institute for Biometrics and Clinical Epidemiology, Charite´, Campus Mitte, Berlin, Germany and 3Department of Hematology and Oncology, University Hospital Mannheim, Mannheim, Germany E26 transforming sequence-related gene (ERG) is a current therapies in acute leukemia patients with poor transcription factor involved in normal hematopoiesis prognosis characterized by high ERG mRNA expression. and is dysregulated in leukemia. ERG mRNA over- Oncogene (2011) 30, 2044–2056; doi:10.1038/onc.2010.582; expression was associated with poor prognosis in a subset published online 17 January 2011 of patients with T-cell acute lymphoblastic leukemia (T-ALL) and acute myeloid leukemia (AML). Herein, a Keywords: ETS-related gene (ERG); WNT11; acute genome-wide screen of ERG target genes was conducted leukemia; ERG target genes; 6-bromoindirubin-3-oxime by chromatin immunoprecipitation-on-chip (ChIP-chip) in (BIO); morphological transformation Jurkat cells. In this screen, 342 significant annotated genes were derived from this global approach. Notably, ERG-enriched targets included WNT signaling genes: WNT11, WNT2, WNT9A, CCND1 and FZD7. Further- more, chromatin immunoprecipitation (ChIP) of normal Introduction and primary leukemia bone marrow material also confirmed WNT11 as a target of ERG in six of seven The erythroblastosis virus E26 transforming sequence patient samples. -

WNT9A Is a Conserved Regulator of Hematopoietic Stem and Progenitor Cell Development
G C A T T A C G G C A T genes Article WNT9A Is a Conserved Regulator of Hematopoietic Stem and Progenitor Cell Development Jenna Richter 1, Edouard G. Stanley 2,3,4, Elizabeth S. Ng 2,3, Andrew G. Elefanty 2,3,4, David Traver 1,* and Karl Willert 1,* ID 1 Department of Cellular and Molecular Medicine, University of California, San Diego, CA 92037, USA; [email protected] 2 Murdoch Childrens Research Institute, The Royal Children’s Hospital, Parkville, VIC 3052, Australia; [email protected] (E.G.S.); [email protected] (E.S.N.); [email protected] (A.G.E.) 3 Department of Anatomy and Developmental Biology, Faculty of Medicine, Nursing and Health Sciences, Monash University, Clayton, VIC 3800, Australia 4 Department of Paediatrics, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Parkville, VIC 3052, Australia * Correspondence: [email protected] (D.T.); [email protected] (K.W.); Tel.: +1-858-822-4593 (D.T.); +1-858-822-3235 (K.W.) Received: 30 November 2017; Accepted: 23 January 2018; Published: 29 January 2018 Abstract: Hematopoietic stem cells (HSCs) differentiate into all cell types of the blood and can be used therapeutically to treat hematopoietic cancers and disorders. Despite decades of research, it is not yet possible to derive therapy-grade HSCs from pluripotent precursors. Analysis of HSC development in model organisms has identified some of the molecular cues that are necessary to instruct hematopoiesis in vivo, including Wnt9A, which is required during an early time window in zebrafish development. -

Cell Regulation
Amphoterin-induced protein 2 (AMIGO2) Q86SJ2 Fibroblast growth factor 21 (FGF21) Q9NSA1 Amyloid beta A4 precursor protein-binding family B Q7Z5R6 Friend leukemia integration 1 transcription factor (FLI1) Q01543 member 1-interacting protein (APBB1IP) Anterior gradient protein 3 (AGR3) Q8TD06 Galectin-7 (LGALS7) P47929 Arylsulfatase B (ARSB) P15848 Gamma-secretase-activating protein (GSAP) A4D1B5 ATP-dependent 6-phosphofructokinase, muscle type (PFKM) P08237 Gastrokine (GKN1) Q9NS71 Bcl-2-like protein 11 isoform BimL (BCL2L11) O43521 GDNF family receptor alpha-2 (GFRA2) O00451 Beta-1,3-galactosyl-O-glycosyl-glycoprotein Q02742 Glucagon (GCG) P01275 beta-1,6-N-acetylglucosaminyltransferase (GCNT1) Biglycan (BGN) P21810 Growth hormone variant (GH2) P01242 Bile salt sulfotransferase (SULT2A1) Q06520 Heparan sulfate glucosamine 3-O-sulfotransferase 3B1 (HS3ST3B1) Q9Y662 Breakpoint cluster region protein (BCR) P11274 Heparan-sulfate 6-O-sulfotransferase 1 (HS6ST1) O60243 Brother of CDO (BOC) Q9BWV1 Immunoglobulin superfamily member 3 (IGSF3) O75054 Calcium/calmodulin-dependent protein kinase kinase 1 (CAMKK1) Q8N5S9 Interleukin-17 receptor B (IL17RB) Q9NRM6 Calsyntenin-3 (CLSTN3) Q9BQT9 Kallikrein-12 (KLK12) Q9UKR0 Cell growth-regulating nucleolar protein (LYAR) Q9NX58 Kazal-type serine protease inhibitor domain-containing protein 1 (KAZALD1) Q96I82 Cellular tumor antigen p53 (TP53) P04637 Leucine-rich repeat neuronal protein 1 (LRRN1) Q6UXK5 Cerebral dopamine neurotrophic factor (CDNF) Q49AH0 Ly6/PLAUR domain-containing protein 1 (LYPD1) Q8N2G4 -

Wnt Signaling Pathway Pharmacogenetics in Non-Small Cell Lung Cancer
The Pharmacogenomics Journal (2014) 14, 509–522 & 2014 Macmillan Publishers Limited All rights reserved 1470-269X/14 www.nature.com/tpj ORIGINAL ARTICLE Wnt signaling pathway pharmacogenetics in non-small cell lung cancer DJ Stewart1, DW Chang2,YYe2, M Spitz2,CLu3, X Shu2, JA Wampfler4, RS Marks5, YI Garces6, P Yang4 and X Wu2 Wingless-type protein (Wnt)/b-catenin pathway alterations in non-small cell lung cancer (NSCLC) are associated with poor prognosis and resistance. In 598 stage III–IV NSCLC patients receiving platinum-based chemotherapy at the MD Anderson Cancer Center (MDACC), we correlated survival with 441 host single-nucleotide polymorphisms (SNPs) in 50 Wnt pathway genes. We then assessed the most significant SNPs in 240 Mayo Clinic patients receiving platinum-based chemotherapy for advanced NSCLC, 127 MDACC patients receiving platinum-based adjuvant chemotherapy and 340 early stage MDACC patients undergoing surgery alone (cohorts 2–4). In multivariate analysis, survival correlates with SNPs for AXIN2 (rs11868547 and rs4541111, of which rs11868547 was assessed in cohorts 2–4), Wnt-5B (rs12819505), CXXC4 (rs4413407) and WIF-1 (rs10878232). Median survival was 19.7, 15.6 and 10.7 months for patients with 1, 2 and 3–5 unfavorable genotypes, respectively (P ¼ 3.8 Â 10 À 9). Survival tree analysis classified patients into two groups (median survival time 11.3 vs 17.3 months, P ¼ 4.7 Â 10 À 8). None of the SNPs achieved significance in cohorts 2–4; however, there was a trend in the same direction as cohort 1 for 3 of the SNPs. Using online databases, we found rs10878232 displayed expression quantitative trait loci correlation with the expression of LEMD3, a neighboring gene previously associated with NSCLC survival. -

Mycobacterium Tuberculosis of Wnt6 Is Expresse
Wnt6 Is Expressed in Granulomatous Lesions of Mycobacterium tuberculosis−Infected Mice and Is Involved in Macrophage Differentiation and Proliferation This information is current as of September 27, 2021. Kolja Schaale, Julius Brandenburg, Andreas Kispert, Michael Leitges, Stefan Ehlers and Norbert Reiling J Immunol 2013; 191:5182-5195; Prepublished online 11 October 2013; doi: 10.4049/jimmunol.1201819 Downloaded from http://www.jimmunol.org/content/191/10/5182 Supplementary http://www.jimmunol.org/content/suppl/2013/10/11/jimmunol.120181 http://www.jimmunol.org/ Material 9.DC1 References This article cites 60 articles, 23 of which you can access for free at: http://www.jimmunol.org/content/191/10/5182.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 27, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights -

Roles of Wnt Pathway Genes Wls, Wnt9a, Wnt5b, Frzb and Gpc4 in Regulating Convergent-Extension During Zebrafish Palate Morphogenesis Lucie Rochard1, Stefanie D
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 2541-2547 doi:10.1242/dev.137000 RESEARCH REPORT Roles of Wnt pathway genes wls, wnt9a, wnt5b, frzb and gpc4 in regulating convergent-extension during zebrafish palate morphogenesis Lucie Rochard1, Stefanie D. Monica2, Irving T. C. Ling1, Yawei Kong1, Sara Roberson3, Richard Harland2, Marnie Halpern3 and Eric C. Liao1,* ABSTRACT Wnts are highly conserved secreted proteins that share a The Wnt signaling pathway is crucial for tissue morphogenesis, common secretion factor, Wntless (Wls), which is a multi-pass participating in cellular behavior changes, notably during the process transmembrane protein that transports Wnt from the Golgi apparatus of convergent-extension. Interactions between Wnt-secreting and to the cell membrane (Bartscherer and Boutros, 2008; Brugmann receiving cells during convergent-extension remain elusive. We et al., 2007; Franch-Marro et al., 2008; Garcia-Castro et al., 2002; investigated the role and genetic interactions of Wnt ligands and Gleason et al., 2006; Harterink and Korswagen, 2012; Lee et al., their trafficking factors Wls, Gpc4 and Frzb in the context of palate 2008; Najdi et al., 2012; Wodarz and Nusse, 1998). Wnts show high morphogenesis in zebrafish. We describe that the chaperon Wls and hydrophobicity, which limits their free diffusion in the extracellular its ligands Wnt9a and Wnt5b are expressed in the ectoderm, whereas space. Extracellular matrix components, such as Glypican (Gpc4), juxtaposed chondrocytes express Frzb and Gpc4. Using wls, gpc4, ensure their diffusion to neighboring cells, while sFRP (Frzb) frzb, wnt9a and wnt5b mutants, we genetically dissected the Wnt enhances diffusion of Wnt by blocking short-range effects and signals operating between secreting ectoderm and receiving increasing long-range gene responses (Cadigan and Peifer, 2009; chondrocytes.