Increased Stability and Activity of Alcohol Oxidase Under High
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-Obese Effects of Chitosan and Psyllium Husk
Progress in Nutrition 2016; Vol. 18, N. 2: 152-160 © Mattioli 1885 Original Article Anti-obese effects of chitosan and psyllium husk containing vitamin C in Sprague-Dawley (SD) rats fed a high fat diet Eun Young Jung1†, Yang Hee Hong2†, Un Jae Chang3, Hyung Joo Suh4 1Department of Home Economics Education, Jeonju University, Jeonju 560-759, Korea; 2Department of Beauty Art, Suwon Women’s University, Suwon 441-748, Korea; 3Department of Food and Nutrition, Dongduk Women’s University, Seoul 136-714, Korea; 4Department of Public Health Science, Graduate School, Korea University, Seoul 07249, Republic of Korea - E-mail: [email protected]; †Eun Young Jung and Yang Hee Hong contributed equally to this study Summary. We investigated whether vitamin C would enhance the anti-obese effects of chitosan and psyllium husk. The study was carried out with male Sprague-Dawley (SD) rats for 6 wk using the following diet groups. Normal diet (control), high fat diet (F-control), 5% chitosan (chitosan-1), 5% chitosan containing 1% vitamin C (chitosan-2), 10% psyllium husk (psyllium-1), and 10% psyllium husk containing 1% vitamin C (psyllium-2). Chitosan-2 (1.5%) showed a significant (p<0.05) decrease in epididymal fat pad weight as compared to F- control (2.3%) and chitosan-1 (2.1%). The epididymal fat pad weight of psyllium-2 (1.9%) was significantly (p<0.05) lower than that of F-control (2.3%). In conclusion, additions of vitamin C to chitosan and psyllium husk may be useful for reducing body fat. Key words: chitosan, psyllium husk, vitamin C, body weight, body fat Introduction models have indicated the efficacy of dietary fiber in regulating body weight (3, 4). -

Starch-Chitosan) Development: Characterization for Packaging Materials
Open Journal of Organic Polymer Materials, 2016, 6, 11-24 Published Online January 2016 in SciRes. http://www.scirp.org/journal/ojopm http://dx.doi.org/10.4236/ojopm.2016.61002 Biodegradable Colour Polymeric Film (Starch-Chitosan) Development: Characterization for Packaging Materials M. Z. I. Mollah1*, N. Akter2, F. B. Quader2, S. Sultana1, R. A. Khan1 1Nuclear and Radiation Chemistry Division, Institute of Nuclear Science and Technology, Bangladesh Atomic Energy Commission, Dhaka, Bangladesh 2Department of Applied Chemistry and Chemical Engineering, University of Dhaka, Dhaka, Bangladesh Received 13 December 2015; accepted 8 January 2016; published 12 January 2016 Copyright © 2016 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Biodegradable starch-based chitosan reinforced composite polymeric films were prepared by casting. The chitosan content in the films was varied from 20% to 80% (w/w). Tensile strength (TS) was improved significantly with the addition of chitosan but elongation at break (EB %) of the composites decreased. Tensile strength of the composites raised more with the addition of the acacia catechu content in the films varied from 0.05% to 0.2% (w/w). The better thermal stability of this prepared film was confirmed by thermo-gravimetric analysis. Structural characterization was done by Fourier transform infrared spectroscopy. Surface morphologies of the composites were examined by scanning electron microscope (SEM) which suggested sufficient homogeniza- tion of starch, chitosan and acacia catechu. Water uptake was found lower for final composites in comparison to starch/chitosan and chitosan films. -

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -

And Honeydew Sugars with Respect to Their Utilization by the Hymenopteran Parasitoid Cotesia Glomerata F.L
Journal of Insect Physiology 47 (2001) 1077–1084 www.elsevier.com/locate/jinsphys A comparison of nectar- and honeydew sugars with respect to their utilization by the hymenopteran parasitoid Cotesia glomerata F.L. Wa¨ckers * Institute of Plant Sciences, Applied Entomology, Swiss Federal Institute of Technology (ETH), 8092 Zurich, Switzerland Received 10 October 2000; received in revised form 12 February 2001; accepted 19 February 2001 Abstract Fourteen naturally occurring sugars were individually tested with respect to their effect on Cotesia glomerata longevity. Parasitoids kept with solutions of either sucrose, glucose and fructose lived for Ͼ30 days. This constitutes a factor 15 increase in life span in comparison to control individuals kept with water only. Stachyose, mannose, melezitose, melibiose, maltose and erlose increased parasitoid longevity by a factor of 11.2–6.9. Solutions of galactose and trehalose had a marginal, but still significant effect. Lactose and raffinose did not raise parasitoid longevity, while rhamnose actually reduced parasitoid survival. In an additional experiment, the relationship between quantity of sugar consumption and longevity was established for all 14 sugars. To study the effect of an unsuitable sugar in sugar mixtures, a range of glucose:rhamnose mixtures was tested. Even at 20% of the sugar mixture rhamnose suppressed the nutritional benefit of the 80% glucose. The nutritional suitability of the sugars shows a positive correlation with the previously reported gustatory response towards the individual sugars. Patterns of sugar utilization are discussed with respect to hydrolytic enzymes and carbohydrate biochemical characteristics. Our findings for C. glomerata are compared to patterns of sugar utilization reported for other species. -

Effect of Intake of Food Hydrocolloids of Bacterial Origin on the Glycemic Response in Humans: Systematic Review and Narrative Synthesis
nutrients Review Effect of Intake of Food Hydrocolloids of Bacterial Origin on the Glycemic Response in Humans: Systematic Review and Narrative Synthesis Norah A. Alshammari 1,2, Moira A. Taylor 3, Rebecca Stevenson 4 , Ourania Gouseti 5, Jaber Alyami 6 , Syahrizal Muttakin 7,8, Serafim Bakalis 5, Alison Lovegrove 9, Guruprasad P. Aithal 2 and Luca Marciani 2,* 1 Department of Clinical Nutrition, College of Applied Medical Sciences, Imam Abdulrahman Bin Faisal University, Dammam 31441, Saudi Arabia; [email protected] 2 Translational Medical Sciences and National Institute for Health Research (NIHR) Nottingham Biomedical Research Centre, Nottingham University Hospitals NHS Trust and University of Nottingham, Nottingham NG7 2UH, UK; [email protected] 3 Division of Physiology, Pharmacology and Neuroscience, School of Life Sciences, Queen’s Medical Centre, National Institute for Health Research (NIHR) Nottingham Biomedical Research Centre, Nottingham NG7 2UH, UK; [email protected] 4 Precision Imaging Beacon, University of Nottingham, Nottingham NG7 2UH, UK; [email protected] 5 Department of Food Science, University of Copenhagen, DK-1958 Copenhagen, Denmark; [email protected] (O.G.); [email protected] (S.B.) 6 Department of Diagnostic Radiology, Faculty of Applied Medical Science, King Abdulaziz University (KAU), Jeddah 21589, Saudi Arabia; [email protected] 7 Indonesian Agency for Agricultural Research and Development, Jakarta 12540, Indonesia; Citation: Alshammari, N.A.; [email protected] Taylor, M.A.; Stevenson, R.; 8 School of Chemical Engineering, University of Birmingham, Birmingham B15 2TT, UK Gouseti, O.; Alyami, J.; Muttakin, S.; 9 Rothamsted Research, Harpenden, Hertfordshire AL5 2JQ, UK; [email protected] Bakalis, S.; Lovegrove, A.; Aithal, G.P.; * Correspondence: [email protected]; Tel.: +44-115-823-1248 Marciani, L. -

Adaptive Laboratory Evolution Enhances Methanol Tolerance and Conversion in Engineered Corynebacterium Glutamicum
ARTICLE https://doi.org/10.1038/s42003-020-0954-9 OPEN Adaptive laboratory evolution enhances methanol tolerance and conversion in engineered Corynebacterium glutamicum Yu Wang 1, Liwen Fan1,2, Philibert Tuyishime1, Jiao Liu1, Kun Zhang1,3, Ning Gao1,3, Zhihui Zhang1,3, ✉ ✉ 1234567890():,; Xiaomeng Ni1, Jinhui Feng1, Qianqian Yuan1, Hongwu Ma1, Ping Zheng1,2,3 , Jibin Sun1,3 & Yanhe Ma1 Synthetic methylotrophy has recently been intensively studied to achieve methanol-based biomanufacturing of fuels and chemicals. However, attempts to engineer platform micro- organisms to utilize methanol mainly focus on enzyme and pathway engineering. Herein, we enhanced methanol bioconversion of synthetic methylotrophs by improving cellular tolerance to methanol. A previously engineered methanol-dependent Corynebacterium glutamicum is subjected to adaptive laboratory evolution with elevated methanol content. Unexpectedly, the evolved strain not only tolerates higher concentrations of methanol but also shows improved growth and methanol utilization. Transcriptome analysis suggests increased methanol con- centrations rebalance methylotrophic metabolism by down-regulating glycolysis and up- regulating amino acid biosynthesis, oxidative phosphorylation, ribosome biosynthesis, and parts of TCA cycle. Mutations in the O-acetyl-L-homoserine sulfhydrylase Cgl0653 catalyzing formation of L-methionine analog from methanol and methanol-induced membrane-bound transporter Cgl0833 are proven crucial for methanol tolerance. This study demonstrates the importance of -

Advances in Chitin/Chitosan Characterization and Applications
Advances in Chitin/Chitosan Characterization and Applications Edited by Marguerite Rinaudo and Francisco M. Goycoolea Printed Edition of the Special Issue Published in Polymers www.mdpi.com/journal/polymers Advances in Chitin/Chitosan Characterization and Applications Advances in Chitin/Chitosan Characterization and Applications Special Issue Editors Marguerite Rinaudo Francisco M. Goycoolea MDPI • Basel • Beijing • Wuhan • Barcelona • Belgrade Special Issue Editors Marguerite Rinaudo Francisco M. Goycoolea University of Grenoble Alpes University of Leeds France UK Editorial Office MDPI St. Alban-Anlage 66 4052 Basel, Switzerland This is a reprint of articles from the Special Issue published online in the open access journal Polymers (ISSN 2073-4360) from 2017 to 2018 (available at: https://www.mdpi.com/journal/polymers/ special issues/chitin chitosan) For citation purposes, cite each article independently as indicated on the article page online and as indicated below: LastName, A.A.; LastName, B.B.; LastName, C.C. Article Title. Journal Name Year, Article Number, Page Range. ISBN 978-3-03897-802-2 (Pbk) ISBN 978-3-03897-803-9 (PDF) c 2019 by the authors. Articles in this book are Open Access and distributed under the Creative Commons Attribution (CC BY) license, which allows users to download, copy and build upon published articles, as long as the author and publisher are properly credited, which ensures maximum dissemination and a wider impact of our publications. The book as a whole is distributed by MDPI under the terms and conditions of the Creative Commons license CC BY-NC-ND. Contents About the Special Issue Editors ..................................... ix Preface to ”Advances in Chitin/Chitosan Characterization and Applications” ......... -

Differential Effects of the Poly (ADP-Ribose)Polymerase (PARP
British Journal of Cancer (2001) 84(1), 106–112 © 2001 Cancer Research Campaign doi: 10.1054/ bjoc.2000.1555, available online at http://www.idealibrary.com on http://www.bjcancer.com Differential effects of the poly (ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro KJ Bowman*, DR Newell, AH Calvert and NJ Curtin Cancer Research Unit, University of Newcastle upon Tyne Medical School, Framlington Place, Newcastle upon Tyne NE2 4HH, UK Summary The potent novel poly(ADP-ribose) polymerase (PARP) inhibitor, NU1025, enhances the cytotoxicity of DNA-methylating agents and ionizing radiation by inhibiting DNA repair. We report here an investigation of the role of PARP in the cellular responses to inhibitors of topoisomerase I and II using NU1025. The cytotoxicity of the topoisomerase I inhibitor, camptothecin, was increased 2.6-fold in L1210 cells by co-incubation with NU1025. Camptothecin-induced DNA strand breaks were also increased 2.5-fold by NU1025 and exposure to camptothecin-activated PARP. In contrast, NU1025 did not increase the DNA strand breakage or cytotoxicity caused by the topoisomerase II inhibitor etoposide. Exposure to etoposide did not activate PARP even at concentrations that caused significant levels of apoptosis. Taken together, these data suggest that potentiation of camptothecin cytotoxicity by NU1025 is a direct result of increased DNA strand breakage, and that activation of PARP by camptothecin-induced DNA damage contributes to its repair and consequently cell survival. However, in L1210 cells at least, it would appear that PARP is not involved in the cellular response to etoposide-mediated DNA damage. -

Electronic Supplementary Information
Electronic Supplementary Material (ESI) for Chemical Science. This journal is © The Royal Society of Chemistry 2019 Electronic Supplementary Information Poly(ionic liquid)s as a Distinct Receptor Material to Create Highly- Integrated Sensing Platform for Efficiently Identifying a Myriad of Saccharides Wanlin Zhang, Yao Li, Yun Liang, Ning Gao, Chengcheng Liu, Shiqiang Wang, Xianpeng Yin, and Guangtao Li* *Corresponding authors: Guangtao Li ([email protected]) S1 Contents 1. Experimental Section (Page S4-S6) Materials and Characterization (Page S4) Experimental Details (Page S4-S6) 2. Figures and Tables (Page S7-S40) Fig. S1 SEM image of silica colloidal crystal spheres and PIL inverse opal spheres. (Page S7) Fig. S2 Adsorption isotherm of PIL inverse opal. (Page S7) Fig. S3 Dynamic mechanical analysis and thermal gravimetric analysis of PIL materials. (Page S7) Fig. S4 Chemical structures of 23 saccharides. (Page S8) Fig. S5 The counteranion exchange of PIL photonic spheres from Br- to DCA. (Page S9) Fig. S6 Reflection and emission spectra of spheres for saccharides. (Page S9) Table S1 The jack-knifed classification on single-sphere array for 23 saccharides. (Page S10) Fig. S7 Lower detection concentration at 10 mM of the single-sphere array. (Page S11) Fig. S8 Lower detection concentration at 1 mM of the single-sphere array. (Page S12) Fig. S9 PIL sphere exhibiting great pH robustness within the biological pH range. (Page S12) Fig. S10 Exploring the tolerance of PIL spheres to different conditions. (Page S13) Fig. S11 Exploring the reusability of PIL spheres. (Page S14) Fig. S12 Responses of spheres to sugar alcohols. (Page S15) Fig. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0028333 A1 Piatesi Et Al
US 20120028333A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0028333 A1 Piatesi et al. (43) Pub. Date: Feb. 2, 2012 (54) USE OF ENZYMES TO REDUCE ALDEHYDES (30) Foreign Application Priority Data FROMALDEHYDE-CONTAINING PRODUCTS Apr. 7, 2009 (EP) .................................. O9157522.5 Publication Classification (76) Inventors: Andrea Piatesi, Mannheim (DE); (51) Int. Cl. Tilo Habicher, Speyer (DE); CI2N 9/02 (2006.01) Michael Bischel, Worms (DE); CI2N I/00 (2006.01) Li-Wen Wang, Mannheim (DE): CI2N 15/63 (2006.01) Jirgen Reichert, Limburgerhof A62D 3/02 (2007.01) (DE); Rainer Packe-Wirth, C7H 2L/04 (2006.01) Trostberg (DE); Kai-Uwe (52) U.S. Cl. ... 435/189: 435/262:536/23.2:435/320.1; Baldenius, Heidelberg (DE); Erich 435/243 Kromm, Weisenheim am Sand (57) ABSTRACT (DE); Stefan Häfner, Speyer (DE); Carsten Schwalb. Mannheim (DE); The invention relates to the use of an enzyme preparation Hans Wolfgang Höffken, which catalyzes the degradation of formaldehyde for reduc Ludwigshafen (DE) ing the formaldehyde content in a formaldehyde-containing formulation. In a preferred embodiment, the enzyme prepa ration contains a formaldehyde dismutase from a Pseudomo (21) Appl. No.: 13/262,662 nas putida Strain. Further, the invention refers to a process for reducing the formaldehyde content in cross-linking agents for textile finishing or in polymer dispersions used, e.g. in con (22) PCT Filed: Mar. 31, 2010 struction chemistry. Further the invention relates to the use of an enzyme preparation which catalyzes the degradation of (86). PCT No.: PCT/EP1OAS4284 aldehydes for reducing the formaldehyde content in an alde hyde-containing formulation. -
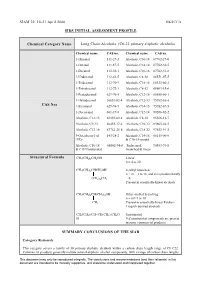
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products. -

Selection of Cryoprotectant in Lyophilization of Progesterone-Loaded Stearic Acid Solid Lipid Nanoparticles
pharmaceutics Article Selection of Cryoprotectant in Lyophilization of Progesterone-Loaded Stearic Acid Solid Lipid Nanoparticles Timothy M. Amis, Jwala Renukuntla, Pradeep Kumar Bolla and Bradley A. Clark * Department of Basic Pharmaceutical Sciences, Fred Wilson School of Pharmacy, High Point University, High Point, NC 27268, USA; [email protected] (T.M.A.); [email protected] (J.R.); [email protected] (P.K.B.) * Correspondence: [email protected]; Tel.: +1-336-841-9665 Received: 18 August 2020; Accepted: 16 September 2020; Published: 19 September 2020 Abstract: Cryoprotectants are often required in lyophilization to reduce or eliminate agglomeration of solute or suspended materials. The aim of this study was to select a cryoprotecting agent and optimize its concentration in a solid lipid nanoparticle formulation. Progesterone-loaded stearic acid solid lipid nanoparticles (SA-P SLNs) were prepared by hot homogenization with high speed mixing and sonication. The stearic acid content was 4.6% w/w and progesterone was 0.46% w/w of the initial formulation. Multiple surfactants were evaluated, and a lecithin and sodium taurocholate system was chosen. Three concentrations of surfactant were then evaluated, and a concentration of 2% w/w was chosen based on particle size, polydispersity, and zeta potential. Agglomeration of SA-P SLNs after lyophilization was observed as measured by increased particle size. Dextran, glycine, mannitol, polyvinylpyrrolidone (PVP), sorbitol, and trehalose were evaluated as cryoprotectants by both an initial freeze–thaw analysis and after lyophilization. Once selected as the cryoprotectant, trehalose was evaluated at 5%, 10%, 15%, and 20% for optimal concentration, with 20% trehalose being finally selected as the level of choice.