Study of Gas-Phase Reactions in Tio2 Production and Functionalised Thin Films Engd Thesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Environmental Protection Agency § 712.30
Environmental Protection Agency § 712.30 (2) A confidentiality statement § 712.30 Chemical lists and reporting signed and dated by an authorized per- periods. son located at the plant site or cor- (a)(1) Persons subject to this subpart porate headquarters of the respondent B must submit a Preliminary Assess- company. ment Information Manufacturer’s Re- (3) The specific chemical name and port for each chemical substance or Chemical Abstracts Service (CAS) Reg- mixture that is listed or designated in istry Number listed in 40 CFR 712.30. this section. (4) The name, company, address, city, State, ZIP code, and telephone number (2) Unless a respondent has already of a person who is submitting the form, prepared a Manufacturer’s Report in which may be a person located at a conformity with conditions set forth in plant site or corporate headquarters paragraph (a)(3) of this section, the in- that will serve as the respondent, and formation in each Manufacturer’s Re- will be able to answer questions about port must cover the respondent’s latest the information submitted by the com- complete corporate fiscal year as of the pany to EPA. A respondent to this sub- effective date. The effective date will part must include the appropriate Dun be 30 days after the FEDERAL REGISTER and Bradstreet Number for each plant publishes a rule amendment making site reported. the substance or mixture subject to (5) The plant site activities, such as this subpart B. the manufacturing of a chemical sub- (3) Persons subject to this subpart B stance, including the total quantity of need not comply with the requirements the chemical substance (in kilograms) of paragraph (a)(2) of this section if imported in bulk during the reporting they meet either one of the following period. -

Crude Ticl4 Purification: a Review of the Current State of the Art and Future Opportunities
Crude TiCl4 purification: a review of the current state of the art and future opportunities L. Hockaday and A. Kale Mintek, Randburg, South Africa Raw titanium tetrachloride (TiCl4) is produced by chlorination of titania feedstock. Prior to its utilization in the manufacture of titanium dioxide pigment or titanium metal, the TiCl4 is purified by adding reagents that react with the impurities in the crude TiCl4, followed by distillation of the mixture. The current state of the art in purification of crude TiCl4 was surveyed. Tests were conducted at the laboratory scale to assess the efficiency of various additives used in the purification process to minimize the vanadium content in the distillate. The effects of various reagents, namely oleic acid, sodium oleate, potassium oleate, and copper on the purification of crude TiCl4 were compared. The purified TiCl4 was water-clear in aspect and analysed < 2 ppm vanadium for all reagents tested. Possible reactions between the impurities in the crude TiCl4 and the reagents are investigated thermodynamically with the FactSage program. INTRODUCTION Titanium tetrachloride (TiCl4) is obtained by the chlorination of a titanium-bearing mineral such as ilmenite and rutile, as well as from titanium slag. The chlorination process usually takes place in a fluidized bed in the presence of chlorine gas and a reducing agent such as petroleum coke, resulting in crude titanium tetrachloride. TiCl4 is an intermediate product in the production of titanium dioxide (TiO2) pigment and titanium metal. The pure form of titanium dioxide is used in the pigment industry for paints, food colouring etc., and titanium metal is widely used in the aerospace, medicine, sport, and semiconductor production industries. -

Thin Films for Smart Windows: Synthesis of Temperature-Responsive Vanadium Oxide for Energy-Efficient Glazing Applications
Thin films for Smart Windows: Synthesis of Temperature-Responsive Vanadium Oxide for Energy-Efficient Glazing Applications Delphine Malardé A thesis submitted to University College London in partial fulfilment of the requirements for the degree of Doctor of Philosophy Department of Chemistry University College London 2019 Declaration ‘I, Delphine Malardé, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis.' Delphine Malardé 2 Abstract The material presented in this thesis highlights different ways of producing temperature-responsive monoclinic VO2 [VO2(M)] for energy-efficient glazing applications, and the improvement of their thermochromic properties. VO2(M) has been widely studied due to its specific thermochromic properties, which make it a promising material to use in home and commercial façade glazing for reducing energy consumptions. VO2(M) thin films were deposited on glass substrate using atmospheric chemical vapour deposition (APCVD) - a well-known deposition process commonly used in industry. The thermochromic properties of the films were optimised, which resulted in a doubling of the visible light transmittance (TLUM) and a fivefold increase in the solar modulation efficiency (ΔTSOL). Further improvements of the thermochromic properties were carried out by the synthesis of VO2-based multilayers. Aerosol-assisted chemical vapour deposition (AACVD) was also used to produce VO2(M) thin films from a newly produced vanadium alkoxide precursor. Furthermore, this precursor showed to be ideal for the formation of V2O3 films with bixbyite structure, which had interesting gas sensing properties. This is the first time that bixbyite-type V2O3 has been synthesised as thin films using AACVD. -

United States Patent Office Patented Sept
3,467,637 United States Patent Office Patented Sept. 16, 1969 1. 2 diene (hereinafter called “bicycloheptadiene'). The reac 3,467,637 tion is carried out using a transition metal catalyst of the LOW MOLECULAR WEIGHT OLEFIN INTER Ziegler type. POLYMERS PREPARED IN THE PRESENCE OF The process described and the copolymers produced in BICYCLO2.2.1]HEPTA-2,5-DIENE Paul J. Prucna, Cheswick, Pa., assignor to PPG Indus the above manner have several distinguishing and ad tries, Inc., a corporation of Pennsylvania Vantageous characteristics. These include: No Drawing. Filled June 1, 1965, Ser. No. 460,551 (a) The process produces low molecular weight co int. C. C08f 15/40, 1/42 s polymer in good yields using easily practised reaction U.S. C. 260-80,78 8 Claims conditions; (b) Varying ratios of ethylene to the other monoolefin O can be employed, permitting variation in product struc ture and properties; ABSTRACT OF THE DISCLOSURE (c) The products are essentially linear and homogene This invention relates to low molecular weight poly ous in structure; mers of ethylene and other alpha monoolefins produced 5 (d) The copolymers are readily dissolved in common by reacting said monomers in the presence of a Ziegler organic Solvents, this being true even with copolymers catalyst and small amounts of bicyclo[2.2.1]hepta-2,5- containing low proportions (e.g., below 20 percent) of diene. The copolymers are useful inter alia in coatings, propylene or other comonomer, whereas ordinarily such adhesives and as tackifying resins. copolymers are quite crystalline and insoluble; and 20 (e) The copolymers are stable and have good aging Stacteristics, attributable to their saturated linear back Oe, This invention relates to low molecular weight co The preferred comonomer with ethylene is propylene, polymers of ethylene and other monoolefins, and, more but other terminal monoolefins such as butylene or iso particularly, to such copolymers produced in the presence 25 butylene can also be utilized. -

Toxicological Review of Vanadium Pentoxide (V2o5)
DRAFT – DO NOT CITE OR QUOTE EPA/635/R-11/004A www.epa.gov/iris TOXICOLOGICAL REVIEW OF VANADIUM PENTOXIDE (V2O5) (CAS No. 1314-62-1) In Support of Summary Information on the Integrated Risk Information System (IRIS) September 2011 NOTICE This document is an External Review draft. This information is distributed solely for the purpose of pre-dissemination peer review under applicable information quality guidelines. It has not been formally disseminated by EPA. It does not represent and should not be construed to represent any Agency determination or policy. It is being circulated for review of its technical accuracy and science policy implications. U.S. Environmental Protection Agency Washington, DC. DISCLAIMER This document is a preliminary draft for review purposes only. This information is distributed solely for the purpose of pre-dissemination peer review under applicable information quality guidelines. It has not been formally disseminated by EPA. It does not represent and should not be construed to represent any Agency determination or policy. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. This document is a draft for review purposes only and does not constitute Agency policy. ii DRAFT – DO NOT CITE OR QUOTE CONTENTS – TOXICOLOGICAL REVIEW OF VANADIUM PENTOXIDE (CAS No. 1314-62-1) LIST OF TABLES ......................................................................................................................... vi LIST OF FIGURES .................................................................................................................... -

Hazardous Waste List (California Code of Regulations, Title 22 Section 66261.126)
Hazardous Waste List (California Code of Regulations, Title 22 Section 66261.126) Appendix X - List of Chemical Names and Common Names for Hazardous Wastes and Hazardous Materials (a) This subdivision sets forth a list of chemicals which create a presumption that a waste is a hazardous waste. If a waste consists of or contains a chemical listed in this subdivision, the waste is presumed to be a hazardous waste Environmental Regulations of CALIFORNIA unless it is determined that the waste is not a hazardous waste pursuant to the procedures set forth in section 66262.11. The hazardous characteristics which serve as a basis for listing the chemicals are indicated in the list as follows: (X) toxic (C) corrosive (I) ignitable (R) reactive * =Extremely Hazardous A chemical denoted with an asterisk is presumed to be an extremely hazardous waste unless it does not exhibit any of the criteria set forth in section 66261.110 and section 66261.113. Trademark chemical names are indicated by all capital letters. 1. Acetaldehyde (X,I) 2. Acetic acid (X,C,I) 3. Acetone, Propanone (I) 4. *Acetone cyanohydrin (X) 5. Acetonitrile (X,I) 6. *2-Acetylaminofluorene, 2-AAF (X) 7. Acetyl benzoyl peroxide (X,I,R) 8. *Acetyl chloride (X,C,R) 9. Acetyl peroxide (X,I,R) 10. Acridine (X) 11. *Acrolein, Aqualin (X,I) 12. *Acrylonitrile (X,I) 13. *Adiponitrile (X) 14. *Aldrin; 1,2,3,4,10,10-Hexachloro-1,4,4a,5,8,8a-hexahydro-1,4,5,8-endo-exodimethanonaphthlene (X) 15. *Alkyl aluminum chloride (C,I,R) 16. *Alkyl aluminum compounds (C,I,R) 17. -

Hazardous Materials Submitted in Duplicate
87510 Federal Register / Vol. 81, No. 233 / Monday, December 5, 2016 / Proposed Rules 40 CFR Part 160 for passengers and crew members. In Avenue SE., 2nd Floor, Washington, DC Environmental protection, addition to harmonization with 20590–0001. international standards, several of the Laboratories, Pesticides and pests, SUPPLEMENTARY INFORMATION: Reporting and recordkeeping proposals in this rule are responsive to requirements. petitions for rulemaking submitted by Table of Contents the regulated community. PHMSA 40 CFR Part 165 invites all interested persons to provide I. Background II. Overview of Proposals in This NPRM Environmental protection, Packaging comments regarding these proposed revisions. A. Transportation by Air Intermediate and containers, Pesticides and pests. Packaging Requirements for Certain Low DATES: Comments must be received by 40 CFR Part 168 and Medium Danger Hazardous February 3, 2017. Materials (P–1637) Environmental protection, ADDRESSES: You may submit comments B. Quantity Limits for Portable Electronic Administrative practice and procedure, by any of the following methods: Medical Devices Carried by Passengers, Advertising, Exports, Labeling, • Federal Rulemaking Portal: http:// Crewmembers, and Air Operators Pesticides and pests, Reporting and www.regulations.gov. Follow the online (P–1649) recordkeeping requirements. instructions for submitting comments. C. NOTOC Harmonization With the ICAO • 40 CFR Part 170 Fax: 1–202–493–2251. TI (P–1487) • Mail: Docket Management System; D. Amendments to Package Inspection Environmental protection, U.S. Department of Transportation, (P–1671) and Securing Requirements Agricultural worker, Employer, Farms, Dockets Operations, M–30, Ground III. Section-by-Section Review Forests, Greenhouses, Nurseries, Floor, Room W12–140, 1200 New Jersey IV. Regulatory Analyses and Notices Pesticide handler, Pesticides, Worker Avenue SE., Washington, DC 20590– A. -

Adeq Minor Source Air Permit
ADEQ MINOR SOURCE AIR PERMIT Permit No. : 0010-AR-10 IS ISSUED TO: Stratcor, Inc. 4285 Malvern Road Hot Springs, AR 71901 Garland County AFIN: 26-00002 THIS PERMIT IS THE ABOVE REFERENCED PERMITTEE’S AUTHORITY TO CONSTRUCT, MODIFY, OPERATE, AND/OR MAINTAIN THE EQUIPMENT AND/OR FACILITY IN THE MANNER AS SET FORTH IN THE DEPARTMENT’S MINOR SOURCE AIR PERMIT AND THE APPLICATION. THIS PERMIT IS ISSUED PURSUANT TO THE PROVISIONS OF THE ARKANSAS WATER AND AIR POLLUTION CONTROL ACT (ARK. CODE ANN. SEC. 8-4-101 ET SEQ.) AND THE REGULATIONS PROMULGATED THEREUNDER, AND IS SUBJECT TO ALL LIMITS AND CONDITIONS CONTAINED HEREIN. Signed: Michael Bonds Date Chief, Air Division Stratcor, Inc. Permit #: 0010-AR-10 AFIN: 26-00002 Table of Contents Section I: FACILITY INFORMATION.................................................................................... 4 Section II: INTRODUCTION...................................................................................................... 5 Summary of Permit Activity.................................................................................................... 5 Process Description................................................................................................................... 5 Regulations .............................................................................................................................. 10 Total Allowable Emissions ..................................................................................................... 10 Section III: PERMIT HISTORY.............................................................................................. -
WO 2017/106591 Al 22 June 2017 (22.06.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/106591 Al 22 June 2017 (22.06.2017) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C07D 307/68 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, PCT/US20 16/067081 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KH, KN, 16 December 2016 (16. 12.2016) KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, (25) Filing Language: English NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, (26) Publication Language: English RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, (30) Priority Data: ZA, ZM, ZW. 14/974,468 18 December 201 5 (18. 12.2015) US (84) Designated States (unless otherwise indicated, for every (71) Applicant: WISCONSIN ALUMNI RESEARCH kind of regional protection available): ARIPO (BW, GH, FOUNDATION [US/US]; 614 Walnut Street, 13th Floor, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, Madison, WI 53726 (US). -
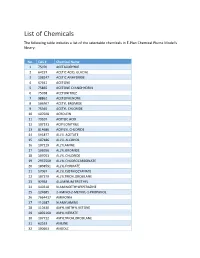
List of Chemicals the Following Table Includes a List of the Selectable Chemicals in E‐Plan Chemical Plume Model's Library
List of Chemicals The following table includes a list of the selectable chemicals in E‐Plan Chemical Plume Model's library: No. CAS # Chemical Name 1 75070 ACETALDEHYDE 2 64197 ACETIC ACID, GLACIAL 3 108247 ACETIC ANHYDRIDE 4 67641 ACETONE 5 75865 ACETONE CYANOHYDRIN 6 75058 ACETONITRILE 7 98862 ACETOPHENONE 8 506967 ACETYL BROMIDE 9 75365 ACETYL CHLORIDE 10 107028 ACROLEIN 11 79107 ACRYLIC ACID 12 107131 ACRYLONITRILE 13 814686 ACRYLYL CHLORIDE 14 591877 ALLYL ACETATE 15 107186 ALLYL ALCOHOL 16 107119 ALLYLAMINE 17 106956 ALLYL BROMIDE 18 107051 ALLYL CHLORIDE 19 2937500 ALLYL CHLOROCARBONATE 20 1838591 ALLYL FORMATE 21 57067 ALLYL ISOTHIOCYANATE 22 107379 ALLYLTRICHLOROSILANE 23 97938 ALUMINUM TRIETHYL 24 140318 N‐AMINOETHYLPIPERAZINE 25 124685 2‐AMINO‐2‐METHYL‐1‐PROPANOL 26 7664417 AMMONIA 27 110587 N‐AMYLAMINE 28 110430 AMYL METHYL KETONE 29 1002160 AMYL NITRATE 30 107722 AMYLTRICHLOROSILANE 31 62533 ANILINE 32 100663 ANISOLE No. CAS # Chemical Name 33 7647189 ANTIMONY PENTACHLORIDE 34 7783702 ANTIMONY PENTAFLUORIDE 35 7784341 ARSENIC TRICHLORIDE 36 7784421 ARSINE 37 71432 BENZENE 38 100470 BENZONITRILE 39 98077 BENZOTRICHLORIDE 40 98088 BENZOTRIFLUORIDE 41 98884 BENZOYL CHLORIDE 42 100469 BENZYLAMINE 43 100390 BENZYL BROMIDE 44 100447 BENZYL CHLORIDE 45 501531 BENZYL CHLOROFORMATE 46 98873 BENZYLIDENE CHLORIDE 47 10294334 BORON TRIBROMIDE 48 10294345 BORON TRICHLORIDE 49 7637072 BORON TRIFLUORIDE 50 109637 BORON TRIFLUORIDE DIETHYL ETHERATE 51 353424 BORON TRIFLUORIDE DIMETHYL ETHERATE 52 7726956 BROMINE 53 13863417 BROMINE CHLORIDE 54 7789302 BROMINE PENTAFLUORIDE 55 7787715 BROMINE TRIFLUORIDE 56 598312 BROMOACETONE 57 108861 BROMOBENZENE 58 109659 1‐BROMOBUTANE 59 353593 BROMOCHLORODIFLUOROMETHANE 60 74975 BROMOCHLOROMETHANE 61 540512 2‐BROMOETHANOL 62 75252 BROMOFORM 63 107824 1‐BROMO‐3‐METHYLBUTANE 64 75638 BROMOTRIFLUOROMETHANE 65 106990 1,3‐BUTADIENE 66 106978 BUTANE 67 106989 1‐BUTENE 68 590181 2‐BUTENE‐CIS 69 624646 2‐BUTENE‐TRANS 70 123864 BUTYL ACETATE 71 105464 SEC‐BUTYL ACETATE No. -

VAPOR-LIQUID EQUILIBRIUM DATA COLLECTION Chemistry Data Series
J. Gmehling U. Onken VAPOR-LIQUID EQUILIBRIUM DATA COLLECTION Halogen, Nitrogen, Sulfur and other Compounds Supplement 1 Chemistry Data Series Vol. I, Part Sa Published by DECH EMA Gesellschaft fUr Chemische Technik und Biotechnologie e. V. Executive Editor: Gerhard Kreysa Die Deutsche Bibliothek - CIP Einheitsaufnahme Chemistry data series I pub!. by DECHEMA, Gesellschaft fOr Chemische Technik und Biotechnologie e.V. Executive ed.: Gerhard Kreysa. - Frankfurt am Main : DECHEMA Teilw. hrsg. von DECHEMA, Deutsche Gesellschaft fi.irChemisches Apparatewesen, Chemische Technik und Biotechnologie e.V. - Teilw. hrsg. von DECHEMA, Deut sche GesellschaftfOr Chemisches Apparatewesen. - Teilw. hrsg. von Dieter Behrens und Reiner Eckermann. - Teilw. hrsg. von Reiner Eckermann und Gerhard Kreysa Vapor liquid equilibrium collection I pub!. by DECHEMA, Gesellschaft fOr Chemi sche Technik und Biotechnologie e. V.. - Frankfurt am Main : DECHEMA (Chemistry data series ; Vol. 1) Teilw. hrsg. von DECHEMA, Deutsche Gesellschaft fOr Chemisches Apparatewesen, Chemische Technik und Biotechnologie e.V. - Teilw. hrsg. von DECHEMA, Deut sche Gesellschaft fOr Chemisches Apparatewesen. - Pt. 8. Halogen, nitrogen, sulfur and other compounds I Jurgen Gmehling ; Ulf On ken a. (Suppl. 1).- 2001 ISBN 3-89746-026-9 © DECHEMA Gesellschaft fOr Chemische Technik und Biotechnologie e. V. Postfach 15 01 04, D-60061 Frankfurt am Main, Germany, 2001 Dieses Werk ist urheberrechtlich geschutzt. Alle Rechte, auch die der Obersetzung, des Nachdrucks und der Vervielfaltigung des Buches oder Teilen daraus sind vorbehalten. Kein Teil des Werkes dart ohne schriftliche Genehmigung der DECHEMA in irgendeiner Form (Fotokopie, Mikrofilm oder einem anderen Verfahren), auch nicht fOr Zwecke der Un terrichtsgestaltung, reproduziert oder unter Verwendung elektronischer Systeme verarbei tet, vervielfaltigt oder verbreitet werden.