Nucleobindin-2: a Novel Regulator of Immune-Metabolic Interactions" (2016)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

Molecular Effects of Isoflavone Supplementation Human Intervention Studies and Quantitative Models for Risk Assessment
Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis committee Promotors Prof. Dr Pieter van ‘t Veer Professor of Nutritional Epidemiology Wageningen University Prof. Dr Evert G. Schouten Emeritus Professor of Epidemiology and Prevention Wageningen University Co-promotors Dr Anouk Geelen Assistant professor, Division of Human Nutrition Wageningen University Dr Lydia A. Afman Assistant professor, Division of Human Nutrition Wageningen University Other members Prof. Dr Jaap Keijer, Wageningen University Dr Hubert P.J.M. Noteborn, Netherlands Food en Consumer Product Safety Authority Prof. Dr Yvonne T. van der Schouw, UMC Utrecht Dr Wendy L. Hall, King’s College London This research was conducted under the auspices of the Graduate School VLAG (Advanced studies in Food Technology, Agrobiotechnology, Nutrition and Health Sciences). Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Friday 20 June 2014 at 13.30 p.m. in the Aula. Vera van der Velpen Molecular effects of isoflavone supplementation: Human intervention studies and quantitative models for risk assessment 154 pages PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in Dutch and English ISBN: 978-94-6173-952-0 ABSTRact Background: Risk assessment can potentially be improved by closely linked experiments in the disciplines of epidemiology and toxicology. -

B Inhibition in a Mouse Model of Chronic Colitis1
The Journal of Immunology Differential Expression of Inflammatory and Fibrogenic Genes and Their Regulation by NF-B Inhibition in a Mouse Model of Chronic Colitis1 Feng Wu and Shukti Chakravarti2 Fibrosis is a major complication of chronic inflammation, as seen in Crohn’s disease and ulcerative colitis, two forms of inflam- matory bowel diseases. To elucidate inflammatory signals that regulate fibrosis, we investigated gene expression changes under- lying chronic inflammation and fibrosis in trinitrobenzene sulfonic acid-induced murine colitis. Six weekly 2,4,6-trinitrobenzene sulfonic acid enemas were given to establish colitis and temporal gene expression patterns were obtained at 6-, 8-, 10-, and 12-wk time points. The 6-wk point, TNBS-w6, was the active, chronic inflammatory stage of the model marked by macrophage, neu- trophil, and CD3؉ and CD4؉ T cell infiltrates in the colon, consistent with the idea that this model is T cell immune response driven. Proinflammatory genes Cxcl1, Ccl2, Il1b, Lcn2, Pla2g2a, Saa3, S100a9, Nos2, Reg2, and Reg3g, and profibrogenic extra- cellular matrix genes Col1a1, Col1a2, Col3a1, and Lum (lumican), encoding a collagen-associated proteoglycan, were up-regulated at the active/chronic inflammatory stages. Rectal administration of the NF-B p65 antisense oligonucleotide reduced but did not abrogate inflammation and fibrosis completely. The antisense oligonucleotide treatment reduced total NF-B by 60% and down- regulated most proinflammatory genes. However, Ccl2, a proinflammatory chemokine known to promote fibrosis, was not down- regulated. Among extracellular matrix gene expressions Lum was suppressed while Col1a1 and Col3a1 were not. Thus, effective treatment of fibrosis in inflammatory bowel disease may require early and complete blockade of NF-B with particular attention to specific proinflammatory and profibrogenic genes that remain active at low levels of NF-B. -

Aprioritized Panel of Candidate Genes
In: Advances in Genetics Research. Volume 9 ISBN: 978-1- 62081-466-6 Editor: Kevin V. Urbano © 2012 Nova Science Publishers, Inc No part of this digital document may be reproduced, stored in a retrieval system or transmitted commercially in any form or by any means. The publisher has taken reasonable care in the preparation of this digital document, but makes no expressed or implied warranty of any kind and assumes no responsibility for any errors or omissions. No liability is assumed for incidental or consequential damages in connection with or arising out of information contained herein. This digital document is sold with the clear understanding that the publisher is not engaged in rendering legal, medical or any other professional services. Chapter 1 A PRIORITIZED PANEL OF CANDIDATE GENES TO BE EXPLORED FOR ASSOCIATIONS WITH THE BLOOD PRESSURE RESPONSE TO EXERCISE Garrett I. Ash,1,Amanda L. Augeri,2John D. Eicher,3 Michael L. Bruneau, Jr.1 and Linda S. Pescatello1 1University of Connecticut, Storrs, CT, US 2Hartford Hospital, Hartford, CT, US 3Yale University School of Medicine, New Haven, CT, US ABSTRACT Introduction. Identifying genetic variants associated with the blood pressure (BP) response to exercise is hindered by the lack of standard criteria for candidate gene selection, underpowered samples, and a limited number of conclusive whole genome studies. We developed a prioritized panel of candidate genetic variants that may increase the likelihood of finding genotype associations with the BP response to exercise when used alone or in conjunction with high throughput genotyping studies. Methods. We searched for candidate genetic variants meeting preestablished criteria using PubMed and published systematic reviews. -

Original Article Nucleobindin 2 (NUCB2) in Renal Cell Carcinoma: a Novel Factor Associated with Tumor Development
Int J Clin Exp Med 2019;12(7):8686-8693 www.ijcem.com /ISSN:1940-5901/IJCEM0090807 Original Article Nucleobindin 2 (NUCB2) in renal cell carcinoma: a novel factor associated with tumor development Ziwei Wei*, Huan Xu*, Qiling Shi*, Long Li, Juan Zhou, Guopeng Yu, Bin Xu, Yushan Liu, Zhong Wang, Wenzhi Li Department of Urology, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, P.R. China. *Equal contributors. Received January 3, 2019; Accepted May 8, 2019; Epub July 15, 2019; Published July 30, 2019 Abstract: This study aimed to examine the expression of NUCB2 in renal cell carcinoma (RCC) tissues and detect its effect on the apoptosis and proliferation of RCC cells both in vivo and vitro. NUCB2 was detected with higher expres- sion in the RCC tissues compared with the adjacent noncancerous tissues by immunohistochemical analysis (P < 0.05). Moreover, the protein levels of NUCB2 was significantly associated with perinephric tissues invasion, cancer- ous thrombus and distant metastasis. NUCB2 knock-down inhibited cell proliferation by arresting the cell cycle at S phase and increased the cell apoptosis detected by CCK-8 and flow cytometry analysis. Finally, the tumor-bearing mice models were constructed through injection of 786-O cells transfected with lentivirus carrying shRNAs targeting NUCB2 or ACTB cDNA plasmids. As expected, the mice in the NUCB2-shRNA group demonstrated a reduced tumor volume and growth rate compared with that in negative control group. This study observed the up-regulated expres- sion of NUCB2 in the ccRCC tissue and 786-O cell line. -
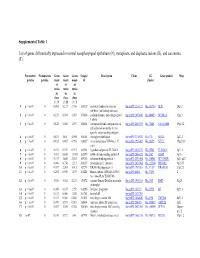
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

A Novel Function of NUCB2 in Promoting the Development and Invasion of Renal Cell Carcinoma
ONCOLOGY LETTERS 15: 2425-2430, 2018 A novel function of NUCB2 in promoting the development and invasion of renal cell carcinoma HUAN XU1*, WENZHI LI1,2*, KAI QI3, JUAN ZHOU1, MENG GU1 and ZHONG WANG1 1Department of Urology, Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200011; Departments of 2Urology and 3Surgery, Linyi People's Hospital, Shandong University, Linyi, Shandong 276003, P.R. China Received April 2, 2016; Accepted June 15, 2017 DOI: 10.3892/ol.2017.7563 Abstract. Previous studies have assessed nucleobindin cell carcinoma. NUCB2 may be an important prognostic 2 (NUCB2) expression in multiple urological cancer cell factor and target in the diagnosis and treatment of human lines and detected its effect on renal cancer cell apoptosis. renal cancer. Additionally, certain reports have indicated a novel function of NUCB2 in promoting invasion in renal cancer. The levels of Introduction NUCB2 expression in different tumor cell lines were detected by quantitative reverse transcription polymerase chain reac- Renal cell carcinoma (RCC) is the most common malignant tion (RT-qPCR). Human NUCB2 and β-actin (ACTB) cDNA neoplasms of the kidney (1). RCC represents 3-4% of adult plasmids were inserted into lentivirus plasmids, which were solid tumors globally, and metastasis is detected in ~30% of then transfected into 786-O cells. Western blotting and patients during initial diagnosis due to the lack of symptoms RT-qPCR were then performed to determine the gene expres- typical of the disease at the early stage (1). Additionally, the sion in NUCB2-knocked down cells. Apoptosis was also 5-year survival of the patients with metastatic RCC has been examined by flow cytometry subsequent to successful trans- identified to be <10% (2). -

Integrated Bioinformatics Analysis Reveals Novel Key Biomarkers and Potential Candidate Small Molecule Drugs in Gestational Diabetes Mellitus
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434569; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Integrated bioinformatics analysis reveals novel key biomarkers and potential candidate small molecule drugs in gestational diabetes mellitus Basavaraj Vastrad1, Chanabasayya Vastrad*2, Anandkumar Tengli3 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. 3. Department of Pharmaceutical Chemistry, JSS College of Pharmacy, Mysuru and JSS Academy of Higher Education & Research, Mysuru, Karnataka, 570015, India * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434569; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Gestational diabetes mellitus (GDM) is one of the metabolic diseases during pregnancy. The identification of the central molecular mechanisms liable for the disease pathogenesis might lead to the advancement of new therapeutic options. The current investigation aimed to identify central differentially expressed genes (DEGs) in GDM. The transcription profiling by array data (E-MTAB-6418) was obtained from the ArrayExpress database. The DEGs between GDM samples and non GDM samples were analyzed with limma package. Gene ontology (GO) and REACTOME enrichment analysis were performed using ToppGene. Then we constructed the protein-protein interaction (PPI) network of DEGs by the Search Tool for the Retrieval of Interacting Genes database (STRING) and module analysis was performed. -

Milger Et Al. Pulmonary CCR2+CD4+ T Cells Are Immune Regulatory And
Milger et al. Pulmonary CCR2+CD4+ T cells are immune regulatory and attenuate lung fibrosis development Supplemental Table S1 List of significantly regulated mRNAs between CCR2+ and CCR2- CD4+ Tcells on Affymetrix Mouse Gene ST 1.0 array. Genewise testing for differential expression by limma t-test and Benjamini-Hochberg multiple testing correction (FDR < 10%). Ratio, significant FDR<10% Probeset Gene symbol or ID Gene Title Entrez rawp BH (1680) 10590631 Ccr2 chemokine (C-C motif) receptor 2 12772 3.27E-09 1.33E-05 9.72 10547590 Klrg1 killer cell lectin-like receptor subfamily G, member 1 50928 1.17E-07 1.23E-04 6.57 10450154 H2-Aa histocompatibility 2, class II antigen A, alpha 14960 2.83E-07 1.71E-04 6.31 10590628 Ccr3 chemokine (C-C motif) receptor 3 12771 1.46E-07 1.30E-04 5.93 10519983 Fgl2 fibrinogen-like protein 2 14190 9.18E-08 1.09E-04 5.49 10349603 Il10 interleukin 10 16153 7.67E-06 1.29E-03 5.28 10590635 Ccr5 chemokine (C-C motif) receptor 5 /// chemokine (C-C motif) receptor 2 12774 5.64E-08 7.64E-05 5.02 10598013 Ccr5 chemokine (C-C motif) receptor 5 /// chemokine (C-C motif) receptor 2 12774 5.64E-08 7.64E-05 5.02 10475517 AA467197 expressed sequence AA467197 /// microRNA 147 433470 7.32E-04 2.68E-02 4.96 10503098 Lyn Yamaguchi sarcoma viral (v-yes-1) oncogene homolog 17096 3.98E-08 6.65E-05 4.89 10345791 Il1rl1 interleukin 1 receptor-like 1 17082 6.25E-08 8.08E-05 4.78 10580077 Rln3 relaxin 3 212108 7.77E-04 2.81E-02 4.77 10523156 Cxcl2 chemokine (C-X-C motif) ligand 2 20310 6.00E-04 2.35E-02 4.55 10456005 Cd74 CD74 antigen -

Universidad Autónoma De Madrid
Universidad Autónoma de Madrid Departamento de Bioquímica Identification of the substrates of the protease MT1-MMP in TNFα- stimulated endothelial cells by quantitative proteomics. Analysis of their potential use as biomarkers in inflammatory bowel disease. Agnieszka A. Koziol Tesis doctoral Madrid, 2013 2 Departamento de Bioquímica Facultad de Medicina Universidad Autónoma de Madrid Identification of the substrates of the protease MT1-MMP in TNFα- stimulated endothelial cells by quantitative proteomics. Analysis of their potential use as biomarkers in inflammatory bowel disease. Memoria presentada por Agnieszka A. Koziol licenciada en Ciencias Biológicas para optar al grado de Doctor. Directora: Dra Alicia García Arroyo Centro Nacional de Investigaciones Cardiovasculares (CNIC) Madrid, 2013 3 4 ACKNOWLEDGEMENTS - ACKNOWLEDGEMENTS - First and foremost I would like to express my sincere gratitude to my supervisor Dr. Alicia García Arroyo for giving me the great opportunity to study under her direction. Her encouragement, guidance and support from the beginning to the end, enabled me to develop and understand the subject. Her feedback to this manuscript, critical reading and corrections was inappreciable to finish this dissertation. Next, I would like to express my gratitude to those who helped me substantially along the way. To all members of MMPs’ lab, for their interest in the project, teaching me during al these years and valuable contribution at the seminary discussion. Without your help it would have not been possible to do some of those experiments. Thank you all for creating a nice atmosphere at work and for being so good friends in private. I would like to also thank all of the collaborators and technical units for their support and professionalism. -

Transcriptomic and Proteomic Analysis of Human Hepatic Stellate Cells Treated with Natural Taurine
1442 MOLECULAR MEDICINE REPORTS 7: 1442-1452, 2013 Transcriptomic and proteomic analysis of human hepatic stellate cells treated with natural taurine JIAN LIANG, XIN DENG, FA‑SHENG WU and YAN‑FANG TANG Ruikang Hospital of Guangxi Traditional Chinese Medical University, Nanning, Guangxi 530011, P.R. China Received September 24, 2012; Accepted February 14, 2013 DOI: 10.3892/mmr.2013.1389 Abstract. The aim of this study was to investigate the Introduction differential expression of genes and proteins between natural taurine (NTau)-treated hepatic stellate cells (HSCs) and Hepatic fibrosis refers to the excessive accumulation of extra- control cells as well as the underlying mechanism of NTau in cellular matrix (ECM) components in the liver. It occurs in inhibiting hepatic fibrosis. A microculture tetrazolium(MTT) most types of chronic liver diseases, including liver cirrhosis, assay was used to analyze the proliferation of NTau‑treated liver failure, and portal hypertension, and often requires liver HSCs. Flow cytometry was performed to compare the transplantation (1). The activation of hepatic stellate cells apoptosis rate between NTau-treated and non-treated HSCs. (HSCs) is an important event by which this cell type, which Proteomic analysis using a combination of 2-dimensional is otherwise quiescent, expresses α-smooth muscle actin gel electrophoresis (2DE) and mass spectrometry (MS) was (α‑SMA), assumes a myofibroblastic phenotype and synthe- conducted to identify the differentially expressed proteins. sizes fibrillar collagens (2,3). Therefore, the inhibition of Microarray analysis was performed to investigate the differ- HSC proliferation, the regulation of the HSC cell cycle, and ential expression of genes and real-time polymerase chain the facilitation of HSC apoptosis are important therapeutic reaction (PCR) was used to validate the results. -

1 Imipramine Treatment and Resiliency Exhibit Similar
Imipramine Treatment and Resiliency Exhibit Similar Chromatin Regulation in the Mouse Nucleus Accumbens in Depression Models Wilkinson et al. Supplemental Material 1. Supplemental Methods 2. Supplemental References for Tables 3. Supplemental Tables S1 – S24 SUPPLEMENTAL TABLE S1: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S2: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S3: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Isolation Model (p<0.001) SUPPLEMENTAL TABLE S4: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Isolation Model (p<0.001) SUPPLEMENTAL TABLE S5: Genes Demonstrating Common Altered Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S6: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S7: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S8: Genes Demonstrating Increased Phospho-CREB Binding in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S9: Genes Demonstrating Decreased Phospho-CREB Binding in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S10: Genes Demonstrating Increased Phospho-CREB Binding in the Social