Modifying the Classical F Test for Microarray Experiments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

Product Description SALSA® MS-MLPA® Probemix ME031-C1 GNAS to Be Used with the MS-MLPA General Protocol
Product description version C1-01; Issued 19 March 2021 Product Description SALSA® MS-MLPA® Probemix ME031-C1 GNAS To be used with the MS-MLPA General Protocol. Version C1 As compared to version B2, probemix completely revised, details are shown in complete product history see page 11. Catalogue numbers: ME031-025R: SALSA MS-MLPA Probemix ME031 GNAS, 25 reactions. ME031-050R: SALSA MS-MLPA Probemix ME031 GNAS, 50 reactions. ME031-100R: SALSA MS-MLPA Probemix ME031 GNAS, 100 reactions. To be used in combination with a SALSA MLPA reagent kit, SALSA HhaI (SMR50) and Coffalyser.Net data analysis software. MLPA reagent kits are either provided with FAM or Cy5.0 dye-labelled PCR primer, suitable for Applied Biosystems and Beckman/SCIEX capillary sequencers, respectively (see www.mrcholland.com). Certificate of Analysis Information regarding storage conditions, quality tests, and a sample electropherogram from the current sales lot is available at www.mrcholland.com. Precautions and warnings For professional use only. Always consult the most recent product description AND the MS-MLPA General Protocol before use: www.mrcholland.com. It is the responsibility of the user to be aware of the latest scientific knowledge of the application before drawing any conclusions from findings generated with this product. This SALSA MS-MLPA probemix is intended for experienced MLPA users only! The exact link between the GNAS complex locus genotype and phenotype is still being investigated. Use of this ME031 GNAS probemix will not always provide you with clear-cut answers and interpretation of results can therefore be complicated. MRC Holland can only provide limited support with interpretation of results obtained with this product, and recommends thoroughly screening any available literature. -

Functional Characterisation of Human Synaptic Genes Expressed in the Drosophila Brain Lysimachos Zografos1,2, Joanne Tang1, Franziska Hesse3, Erich E
© 2016. Published by The Company of Biologists Ltd | Biology Open (2016) 5, 662-667 doi:10.1242/bio.016261 METHODS & TECHNIQUES Functional characterisation of human synaptic genes expressed in the Drosophila brain Lysimachos Zografos1,2, Joanne Tang1, Franziska Hesse3, Erich E. Wanker3, Ka Wan Li4, August B. Smit4, R. Wayne Davies1,5 and J. Douglas Armstrong1,5,* ABSTRACT systems biology approaches are likely to be the best route to unlock a Drosophila melanogaster is an established and versatile model new generation of neuroscience research and CNS drug organism. Here we describe and make available a collection of development that society so urgently demands (Catalá-López transgenic Drosophila strains expressing human synaptic genes. The et al., 2013). Yet these modelling type approaches also need fast, collection can be used to study and characterise human synaptic tractable in vivo models for validation. genes and their interactions and as controls for mutant studies. It was More than 100 years after the discovery of the white gene in generated in a way that allows the easy addition of new strains, as well Drosophila melanogaster, the common fruit fly remains a key tool as their combination. In order to highlight the potential value of the for the study of neuroscience and neurobiology. The fruit fly collection for the characterisation of human synaptic genes we also genome is well annotated and there is a vast genetic manipulation use two assays, investigating any gain-of-function motor and/or toolkit available. This allows interventions such as high throughput cognitive phenotypes in the strains in this collection. Using these cloning (Bischof et al., 2013; Wang et al., 2012) and the precise assays we show that among the strains made there are both types of insertion of transgenes in the genome (Groth et al., 2004; Venken gain-of-function phenotypes investigated. -

Molecular Effects of Isoflavone Supplementation Human Intervention Studies and Quantitative Models for Risk Assessment
Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis committee Promotors Prof. Dr Pieter van ‘t Veer Professor of Nutritional Epidemiology Wageningen University Prof. Dr Evert G. Schouten Emeritus Professor of Epidemiology and Prevention Wageningen University Co-promotors Dr Anouk Geelen Assistant professor, Division of Human Nutrition Wageningen University Dr Lydia A. Afman Assistant professor, Division of Human Nutrition Wageningen University Other members Prof. Dr Jaap Keijer, Wageningen University Dr Hubert P.J.M. Noteborn, Netherlands Food en Consumer Product Safety Authority Prof. Dr Yvonne T. van der Schouw, UMC Utrecht Dr Wendy L. Hall, King’s College London This research was conducted under the auspices of the Graduate School VLAG (Advanced studies in Food Technology, Agrobiotechnology, Nutrition and Health Sciences). Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Friday 20 June 2014 at 13.30 p.m. in the Aula. Vera van der Velpen Molecular effects of isoflavone supplementation: Human intervention studies and quantitative models for risk assessment 154 pages PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in Dutch and English ISBN: 978-94-6173-952-0 ABSTRact Background: Risk assessment can potentially be improved by closely linked experiments in the disciplines of epidemiology and toxicology. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal -

B Inhibition in a Mouse Model of Chronic Colitis1
The Journal of Immunology Differential Expression of Inflammatory and Fibrogenic Genes and Their Regulation by NF-B Inhibition in a Mouse Model of Chronic Colitis1 Feng Wu and Shukti Chakravarti2 Fibrosis is a major complication of chronic inflammation, as seen in Crohn’s disease and ulcerative colitis, two forms of inflam- matory bowel diseases. To elucidate inflammatory signals that regulate fibrosis, we investigated gene expression changes under- lying chronic inflammation and fibrosis in trinitrobenzene sulfonic acid-induced murine colitis. Six weekly 2,4,6-trinitrobenzene sulfonic acid enemas were given to establish colitis and temporal gene expression patterns were obtained at 6-, 8-, 10-, and 12-wk time points. The 6-wk point, TNBS-w6, was the active, chronic inflammatory stage of the model marked by macrophage, neu- trophil, and CD3؉ and CD4؉ T cell infiltrates in the colon, consistent with the idea that this model is T cell immune response driven. Proinflammatory genes Cxcl1, Ccl2, Il1b, Lcn2, Pla2g2a, Saa3, S100a9, Nos2, Reg2, and Reg3g, and profibrogenic extra- cellular matrix genes Col1a1, Col1a2, Col3a1, and Lum (lumican), encoding a collagen-associated proteoglycan, were up-regulated at the active/chronic inflammatory stages. Rectal administration of the NF-B p65 antisense oligonucleotide reduced but did not abrogate inflammation and fibrosis completely. The antisense oligonucleotide treatment reduced total NF-B by 60% and down- regulated most proinflammatory genes. However, Ccl2, a proinflammatory chemokine known to promote fibrosis, was not down- regulated. Among extracellular matrix gene expressions Lum was suppressed while Col1a1 and Col3a1 were not. Thus, effective treatment of fibrosis in inflammatory bowel disease may require early and complete blockade of NF-B with particular attention to specific proinflammatory and profibrogenic genes that remain active at low levels of NF-B. -

Identification of Key Genes and Pathways for Alzheimer's Disease
Biophys Rep 2019, 5(2):98–109 https://doi.org/10.1007/s41048-019-0086-2 Biophysics Reports RESEARCH ARTICLE Identification of key genes and pathways for Alzheimer’s disease via combined analysis of genome-wide expression profiling in the hippocampus Mengsi Wu1,2, Kechi Fang1, Weixiao Wang1,2, Wei Lin1,2, Liyuan Guo1,2&, Jing Wang1,2& 1 CAS Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences, Beijing 100101, China 2 Department of Psychology, University of Chinese Academy of Sciences, Beijing 10049, China Received: 8 August 2018 / Accepted: 17 January 2019 / Published online: 20 April 2019 Abstract In this study, combined analysis of expression profiling in the hippocampus of 76 patients with Alz- heimer’s disease (AD) and 40 healthy controls was performed. The effects of covariates (including age, gender, postmortem interval, and batch effect) were controlled, and differentially expressed genes (DEGs) were identified using a linear mixed-effects model. To explore the biological processes, func- tional pathway enrichment and protein–protein interaction (PPI) network analyses were performed on the DEGs. The extended genes with PPI to the DEGs were obtained. Finally, the DEGs and the extended genes were ranked using the convergent functional genomics method. Eighty DEGs with q \ 0.1, including 67 downregulated and 13 upregulated genes, were identified. In the pathway enrichment analysis, the 80 DEGs were significantly enriched in one Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, GABAergic synapses, and 22 Gene Ontology terms. These genes were mainly involved in neuron, synaptic signaling and transmission, and vesicle metabolism. These processes are all linked to the pathological features of AD, demonstrating that the GABAergic system, neurons, and synaptic function might be affected in AD. -

Impaired Iloprost-Induced Platelet Inhibition and Phosphoproteome Changes in Patients with Confirmed Pseudohypoparathyroidism Ty
www.nature.com/scientificreports OPEN Impaired iloprost‑induced platelet inhibition and phosphoproteome changes in patients with confrmed pseudohypoparathyroidism type Ia, linked to genetic mutations in GNAS Frauke Swieringa1,2, Fiorella A. Solari1,9, Oliver Pagel1,9, Florian Beck1, Jingnan Huang1,2, Marion A. H. Feijge2, Kerstin Jurk4, Irene M. L. W. Körver‑Keularts5, Nadine J. A. Mattheij2, Jörg Faber3, Joachim Pohlenz3, Alexandra Russo3, Connie T. R. M. Stumpel5,6, Dirk E. Schrander7, Barbara Zieger8, Paola E. J. van der Meijden2, René P. Zahedi1, Albert Sickmann1 & Johan W. M. Heemskerk2* Patients diagnosed with pseudohypoparathyroidism type Ia (PHP Ia) sufer from hormonal resistance and abnormal postural features, in a condition classifed as Albright hereditary osteodystrophy (AHO) syndrome. This syndrome is linked to a maternally inherited mutation in the GNAS complex locus, encoding for the GTPase subunit Gsα. Here, we investigated how platelet phenotype and omics analysis can assist in the often difcult diagnosis. By coupling to the IP receptor, Gsα induces platelet inhibition via adenylyl cyclase and cAMP-dependent protein kinase A (PKA). In platelets from seven patients with suspected AHO, one of the largest cohorts examined, we studied the PKA-induced phenotypic changes. Five patients with a confrmed GNAS mutation, displayed impairments in Gsα- dependent VASP phosphorylation, aggregation, and microfuidic thrombus formation. Analysis of the platelet phosphoproteome revealed 2,516 phosphorylation sites, of which 453 were regulated by Gsα-PKA. Common changes in the patients were: (1) a joint panel of upregulated and downregulated phosphopeptides; (2) overall PKA dependency of the upregulated phosphopeptides; (3) links to key platelet function pathways. In one patient with GNAS mutation, diagnosed as non‑AHO, the changes in platelet phosphoproteome were reversed. -

Aprioritized Panel of Candidate Genes
In: Advances in Genetics Research. Volume 9 ISBN: 978-1- 62081-466-6 Editor: Kevin V. Urbano © 2012 Nova Science Publishers, Inc No part of this digital document may be reproduced, stored in a retrieval system or transmitted commercially in any form or by any means. The publisher has taken reasonable care in the preparation of this digital document, but makes no expressed or implied warranty of any kind and assumes no responsibility for any errors or omissions. No liability is assumed for incidental or consequential damages in connection with or arising out of information contained herein. This digital document is sold with the clear understanding that the publisher is not engaged in rendering legal, medical or any other professional services. Chapter 1 A PRIORITIZED PANEL OF CANDIDATE GENES TO BE EXPLORED FOR ASSOCIATIONS WITH THE BLOOD PRESSURE RESPONSE TO EXERCISE Garrett I. Ash,1,Amanda L. Augeri,2John D. Eicher,3 Michael L. Bruneau, Jr.1 and Linda S. Pescatello1 1University of Connecticut, Storrs, CT, US 2Hartford Hospital, Hartford, CT, US 3Yale University School of Medicine, New Haven, CT, US ABSTRACT Introduction. Identifying genetic variants associated with the blood pressure (BP) response to exercise is hindered by the lack of standard criteria for candidate gene selection, underpowered samples, and a limited number of conclusive whole genome studies. We developed a prioritized panel of candidate genetic variants that may increase the likelihood of finding genotype associations with the BP response to exercise when used alone or in conjunction with high throughput genotyping studies. Methods. We searched for candidate genetic variants meeting preestablished criteria using PubMed and published systematic reviews. -

Epigenetic Modifications to Cytosine and Alzheimer's Disease
University of Kentucky UKnowledge Theses and Dissertations--Chemistry Chemistry 2017 EPIGENETIC MODIFICATIONS TO CYTOSINE AND ALZHEIMER’S DISEASE: A QUANTITATIVE ANALYSIS OF POST-MORTEM TISSUE Elizabeth M. Ellison University of Kentucky, [email protected] Digital Object Identifier: https://doi.org/10.13023/ETD.2017.398 Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Ellison, Elizabeth M., "EPIGENETIC MODIFICATIONS TO CYTOSINE AND ALZHEIMER’S DISEASE: A QUANTITATIVE ANALYSIS OF POST-MORTEM TISSUE" (2017). Theses and Dissertations--Chemistry. 86. https://uknowledge.uky.edu/chemistry_etds/86 This Doctoral Dissertation is brought to you for free and open access by the Chemistry at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Chemistry by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

Original Article Nucleobindin 2 (NUCB2) in Renal Cell Carcinoma: a Novel Factor Associated with Tumor Development
Int J Clin Exp Med 2019;12(7):8686-8693 www.ijcem.com /ISSN:1940-5901/IJCEM0090807 Original Article Nucleobindin 2 (NUCB2) in renal cell carcinoma: a novel factor associated with tumor development Ziwei Wei*, Huan Xu*, Qiling Shi*, Long Li, Juan Zhou, Guopeng Yu, Bin Xu, Yushan Liu, Zhong Wang, Wenzhi Li Department of Urology, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, P.R. China. *Equal contributors. Received January 3, 2019; Accepted May 8, 2019; Epub July 15, 2019; Published July 30, 2019 Abstract: This study aimed to examine the expression of NUCB2 in renal cell carcinoma (RCC) tissues and detect its effect on the apoptosis and proliferation of RCC cells both in vivo and vitro. NUCB2 was detected with higher expres- sion in the RCC tissues compared with the adjacent noncancerous tissues by immunohistochemical analysis (P < 0.05). Moreover, the protein levels of NUCB2 was significantly associated with perinephric tissues invasion, cancer- ous thrombus and distant metastasis. NUCB2 knock-down inhibited cell proliferation by arresting the cell cycle at S phase and increased the cell apoptosis detected by CCK-8 and flow cytometry analysis. Finally, the tumor-bearing mice models were constructed through injection of 786-O cells transfected with lentivirus carrying shRNAs targeting NUCB2 or ACTB cDNA plasmids. As expected, the mice in the NUCB2-shRNA group demonstrated a reduced tumor volume and growth rate compared with that in negative control group. This study observed the up-regulated expres- sion of NUCB2 in the ccRCC tissue and 786-O cell line. -
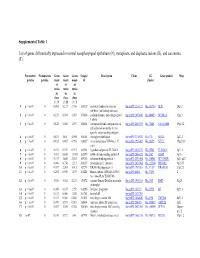
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146.