Impaired Iloprost-Induced Platelet Inhibition and Phosphoproteome Changes in Patients with Confirmed Pseudohypoparathyroidism Ty
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Product Description SALSA® MS-MLPA® Probemix ME031-C1 GNAS to Be Used with the MS-MLPA General Protocol
Product description version C1-01; Issued 19 March 2021 Product Description SALSA® MS-MLPA® Probemix ME031-C1 GNAS To be used with the MS-MLPA General Protocol. Version C1 As compared to version B2, probemix completely revised, details are shown in complete product history see page 11. Catalogue numbers: ME031-025R: SALSA MS-MLPA Probemix ME031 GNAS, 25 reactions. ME031-050R: SALSA MS-MLPA Probemix ME031 GNAS, 50 reactions. ME031-100R: SALSA MS-MLPA Probemix ME031 GNAS, 100 reactions. To be used in combination with a SALSA MLPA reagent kit, SALSA HhaI (SMR50) and Coffalyser.Net data analysis software. MLPA reagent kits are either provided with FAM or Cy5.0 dye-labelled PCR primer, suitable for Applied Biosystems and Beckman/SCIEX capillary sequencers, respectively (see www.mrcholland.com). Certificate of Analysis Information regarding storage conditions, quality tests, and a sample electropherogram from the current sales lot is available at www.mrcholland.com. Precautions and warnings For professional use only. Always consult the most recent product description AND the MS-MLPA General Protocol before use: www.mrcholland.com. It is the responsibility of the user to be aware of the latest scientific knowledge of the application before drawing any conclusions from findings generated with this product. This SALSA MS-MLPA probemix is intended for experienced MLPA users only! The exact link between the GNAS complex locus genotype and phenotype is still being investigated. Use of this ME031 GNAS probemix will not always provide you with clear-cut answers and interpretation of results can therefore be complicated. MRC Holland can only provide limited support with interpretation of results obtained with this product, and recommends thoroughly screening any available literature. -

Genome-Wide Association Study Identifies Loci for Arterial Stiffness
www.nature.com/scientificreports OPEN Genome-wide association study identifes loci for arterial stifness index in 127,121 UK Biobank Received: 1 February 2019 Accepted: 5 June 2019 participants Published: xx xx xxxx Kenneth Fung1, Julia Ramírez 2, Helen R. Warren2,3, Nay Aung 1, Aaron M. Lee1, Evan Tzanis2,3, Stefen E. Petersen 1,3 & Patricia B. Munroe2,3 Arterial stifness index (ASI) is a non-invasive measure of arterial stifness using infra-red fnger sensors (photoplethysmography). It is a well-suited measure for large populations as it is relatively inexpensive to perform, and data can be acquired within seconds. These features raise interest in using ASI as a tool to estimate cardiovascular disease risk as prior work demonstrates increased arterial stifness is associated with elevated systolic blood pressure, and ASI is predictive of cardiovascular disease and mortality. We conducted genome-wide association studies (GWASs) for ASI in 127,121 UK Biobank participants of European-ancestry. Our primary analyses identifed variants at four loci reaching genome-wide signifcance (P < 5 × 10−8): TEX41 (rs1006923; P = 5.3 × 10−12), FOXO1 (rs7331212; P = 2.2 × 10−11), C1orf21 (rs1930290, P = 1.1 × 10−8) and MRVI1 (rs10840457, P = 3.4 × 10−8). Gene- based testing revealed three signifcant genes, the most signifcant gene was COL4A2 (P = 1.41 × 10−8) encoding type IV collagen. Other candidate genes at associated loci were also involved in smooth muscle tone regulation. Our fndings provide new information for understanding the development of arterial stifness. Arterial stifness measures have been reported as independent markers of vascular ageing1,2, hypertension3,4, car- diovascular disease (CVD)5,6 and mortality6,7. -

Functional Characterisation of Human Synaptic Genes Expressed in the Drosophila Brain Lysimachos Zografos1,2, Joanne Tang1, Franziska Hesse3, Erich E
© 2016. Published by The Company of Biologists Ltd | Biology Open (2016) 5, 662-667 doi:10.1242/bio.016261 METHODS & TECHNIQUES Functional characterisation of human synaptic genes expressed in the Drosophila brain Lysimachos Zografos1,2, Joanne Tang1, Franziska Hesse3, Erich E. Wanker3, Ka Wan Li4, August B. Smit4, R. Wayne Davies1,5 and J. Douglas Armstrong1,5,* ABSTRACT systems biology approaches are likely to be the best route to unlock a Drosophila melanogaster is an established and versatile model new generation of neuroscience research and CNS drug organism. Here we describe and make available a collection of development that society so urgently demands (Catalá-López transgenic Drosophila strains expressing human synaptic genes. The et al., 2013). Yet these modelling type approaches also need fast, collection can be used to study and characterise human synaptic tractable in vivo models for validation. genes and their interactions and as controls for mutant studies. It was More than 100 years after the discovery of the white gene in generated in a way that allows the easy addition of new strains, as well Drosophila melanogaster, the common fruit fly remains a key tool as their combination. In order to highlight the potential value of the for the study of neuroscience and neurobiology. The fruit fly collection for the characterisation of human synaptic genes we also genome is well annotated and there is a vast genetic manipulation use two assays, investigating any gain-of-function motor and/or toolkit available. This allows interventions such as high throughput cognitive phenotypes in the strains in this collection. Using these cloning (Bischof et al., 2013; Wang et al., 2012) and the precise assays we show that among the strains made there are both types of insertion of transgenes in the genome (Groth et al., 2004; Venken gain-of-function phenotypes investigated. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Identification of Key Genes and Pathways for Alzheimer's Disease
Biophys Rep 2019, 5(2):98–109 https://doi.org/10.1007/s41048-019-0086-2 Biophysics Reports RESEARCH ARTICLE Identification of key genes and pathways for Alzheimer’s disease via combined analysis of genome-wide expression profiling in the hippocampus Mengsi Wu1,2, Kechi Fang1, Weixiao Wang1,2, Wei Lin1,2, Liyuan Guo1,2&, Jing Wang1,2& 1 CAS Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences, Beijing 100101, China 2 Department of Psychology, University of Chinese Academy of Sciences, Beijing 10049, China Received: 8 August 2018 / Accepted: 17 January 2019 / Published online: 20 April 2019 Abstract In this study, combined analysis of expression profiling in the hippocampus of 76 patients with Alz- heimer’s disease (AD) and 40 healthy controls was performed. The effects of covariates (including age, gender, postmortem interval, and batch effect) were controlled, and differentially expressed genes (DEGs) were identified using a linear mixed-effects model. To explore the biological processes, func- tional pathway enrichment and protein–protein interaction (PPI) network analyses were performed on the DEGs. The extended genes with PPI to the DEGs were obtained. Finally, the DEGs and the extended genes were ranked using the convergent functional genomics method. Eighty DEGs with q \ 0.1, including 67 downregulated and 13 upregulated genes, were identified. In the pathway enrichment analysis, the 80 DEGs were significantly enriched in one Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, GABAergic synapses, and 22 Gene Ontology terms. These genes were mainly involved in neuron, synaptic signaling and transmission, and vesicle metabolism. These processes are all linked to the pathological features of AD, demonstrating that the GABAergic system, neurons, and synaptic function might be affected in AD. -

Epigenetic Modifications to Cytosine and Alzheimer's Disease
University of Kentucky UKnowledge Theses and Dissertations--Chemistry Chemistry 2017 EPIGENETIC MODIFICATIONS TO CYTOSINE AND ALZHEIMER’S DISEASE: A QUANTITATIVE ANALYSIS OF POST-MORTEM TISSUE Elizabeth M. Ellison University of Kentucky, [email protected] Digital Object Identifier: https://doi.org/10.13023/ETD.2017.398 Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Ellison, Elizabeth M., "EPIGENETIC MODIFICATIONS TO CYTOSINE AND ALZHEIMER’S DISEASE: A QUANTITATIVE ANALYSIS OF POST-MORTEM TISSUE" (2017). Theses and Dissertations--Chemistry. 86. https://uknowledge.uky.edu/chemistry_etds/86 This Doctoral Dissertation is brought to you for free and open access by the Chemistry at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Chemistry by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

Germline-Derived Gain-Of-Function Variants of Gsa-Coding GNAS Gene Identified in Nephrogenic Syndrome of Inappropriate Antidiuresis
CLINICAL RESEARCH www.jasn.org Germline-Derived Gain-of-Function Variants of Gsa-Coding GNAS Gene Identified in Nephrogenic Syndrome of Inappropriate Antidiuresis Mami Miyado,1 Maki Fukami,1 Shuji Takada,2 Miho Terao,2 Kazuhiko Nakabayashi,3 Kenichiro Hata,3 Yoichi Matsubara,4 Yoko Tanaka ,5 Goro Sasaki,5 Keisuke Nagasaki ,6 Masaaki Shiina,7 Kazuhiro Ogata,7 Youhei Masunaga,8 Hirotomo Saitsu,9 and Tsutomu Ogata 1,8 Departments of 1Molecular Endocrinology, 2Systems BioMedicine, 3Maternal-Fetal Biology and 4Head Office, National Research Institute for Child Health and Development, Tokyo, Japan; 5Department of Pediatrics, Tokyo Dental College, Ichikawa General Hospital, Ichikawa, Japan; 6Department of Homeostatic Regulation and Development, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan; 7Department of Biochemistry, Yokohama City University Graduate School of Medicine, Yokohama, Japan; and Departments of 8Pediatrics and 9Biochemistry, Hamamatsu University School of Medicine, Hamamatsu, Japan ABSTRACT Background The stimulatory G-protein a-subunit encoded by GNAS exons 1–13 (GNAS-Gsa)mediates signal transduction of multiple G protein–coupled receptors, including arginine vasopressin receptor 2 (AVPR2). Various germline-derived loss-of-function GNAS-Gsa variants of maternal and paternal origin have been found in pseudohypoparathyroidism type Ia and pseudopseudohypoparathyroidism, respec- tively. Specific somatic gain-of-function GNAS-Gsa variants have been detected in McCune–Albright syndrome and may result in phosphate wasting. However, no germline-derived gain-of-function variant has been identified, implying that such a variant causes embryonic lethality. Methods We performed whole-exome sequencing in two families with dominantly inherited nephrogenic syndrome of inappropriate antidiuresis (NSIAD) as a salient phenotype after excluding a gain-of-function variant of AVPR2 and functional studies for identified variants. -
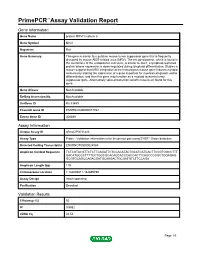
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name protein MRVI1 isoform b Gene Symbol Mrvi1 Organism Rat Gene Summary This gene is similar to a putative mouse tumor suppressor gene that is frequently disrupted by mouse AIDS-related virus (MRV). The encoded protein, which is found in the membrane of the endoplasmic reticulum, is similar to Jaw1, a lymphoid-restricted protein whose expression is down-regulated during lymphoid differentiation. Studies in mouse suggest that MRV integration at the homologous mouse gene induces myeloid leukemia by altering the expression of a gene important for myeloid cell growth and/or differentiation, and thus this gene may function as a myeloid leukemia tumor suppressor gene. Alternatively spliced transcript variants have been found for this gene. Gene Aliases Not Available RefSeq Accession No. Not Available UniGene ID Rn.33889 Ensembl Gene ID ENSRNOG00000017767 Entrez Gene ID 308899 Assay Information Unique Assay ID qRnoCIP0031443 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSRNOT00000024060 Amplicon Context Sequence TCTCGTACGTTCTCTTCAGATTCTCCACATACTGCATCATCACTTCCGTGGCCTTT GACATGCGCTTTTCCTGGCGCACAGCACCCACCACTTCAGCCCGGCTGGAGAG GCGTGCAGCCAGACGATGCAAGACTGCGATGTCTTCCAGA Amplicon Length (bp) 119 Chromosome Location 1:182696811-182699799 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 95 R2 0.9982 cDNA Cq 24.54 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 88 gDNA Cq Specificity -

Mrvil, a Common MRV Integration Site in BXH2 Myeloid Leukemias, Encodes a Protein with Homology to a Lymphoid-Restricted Membrane Protein Jaw1
Oncogene (1999) 18, 2069 ± 2084 ã 1999 Stockton Press All rights reserved 0950 ± 9232/99 $12.00 http://www.stockton-press.co.uk/onc Mrvil, a common MRV integration site in BXH2 myeloid leukemias, encodes a protein with homology to a lymphoid-restricted membrane protein Jaw1 John D Shaughnessy Jr*,1, David A Largaespada2, Erming Tian1, Colin F Fletcher3, Brian C Cho4, Paresh Vyas5, Nancy A Jenkins3 and Neal G Copeland3 1Division of Hematology and Oncology, Department of Medicine, University of Arkansas for Medical Sciences, Little Rock, Arkansas, AR 72205, USA; 2Department of Laboratory Medicine and Pathology, University of Minnesota Cancer Center, Minneapolis, Minnesota, MN 55455, USA; 3Mammalian Genetics Laboratory, ABL-Basic Research Program, NCI-Frederick Cancer Research and Development Center, Frederick, Maryland, MD 21702, USA; 4Department of Microbiology and Immunology, Thomas Jeerson University Cancer Institute, Philadelphia, Pennsylvania, PA 19107, USA; 5Department of Hematology and Oncology, Children's Hospital, Boston, Massachusetts, USA Ecotropic MuLVs induce myeloid leukemia in BXH2 Introduction mice by insertional mutagenesis of cellular proto- oncogenes or tumor suppressor genes. Disease genes BXH2 mice represent an important model for the can thus be identi®ed by viral tagging as common sites of identi®cation of myeloid leukemia disease genes. Not viral integration in BXH2 leukemias. Previous studies only do these mice have the highest spontaneous showed that a frequent common integration site in BXH2 incidence of myeloid leukemia of any known inbred leukemias is the Nf1 tumor suppressor gene. Unexpect- mouse strain, but the leukemias in these mice are edly, about half of the viral integrations at Nf1 retrovirally-induced and the proviruses in the tumors represented a previously undiscovered defective none- can thus be used as insertional tags to identify the cotropic virus, termed MRV. -

Description: Uniprot:Q13976 Alternative Names: Specificity
TD2677 PRKG1 Antibody Order 021-34695924 [email protected] Support 400-6123-828 50ul [email protected] 100 uL √ √ Web www.ab-mart.com.cn Description: Serine/threonine protein kinase that acts as key mediator of the nitric oxide (NO)/cGMP signaling pathway. GMP binding activates PRKG1, which phosphorylates serines and threonines on many cellular proteins. Numerous protein targets for PRKG1 phosphorylation are implicated in modulating cellular calcium, but the contribution of each of these targets may vary substantially among cell types. Proteins that are phosphorylated by PRKG1 regulate platelet activation and adhesion, smooth muscle contraction, cardiac function, gene expression, feedback of the NO-signaling pathway, and other processes involved in several aspects of the CNS like axon guidance, hippocampal and cerebellar learning, circadian rhythm and nociception. Smooth muscle relaxation is mediated through lowering of intracellular free calcium, by desensitization of contractile proteins to calcium, and by decrease in the contractile state of smooth muscle or in platelet activation. Regulates intracellular calcium levels via several pathways: phosphorylates MRVI1/IRAG and inhibits IP3-induced Ca(2+) release from intracellular stores, phosphorylation of KCNMA1 (BKCa) channels decreases intracellular Ca(2+) levels, which leads to increased opening of this channel. PRKG1 phosphorylates the canonical transient receptor potential channel (TRPC) family which inactivates the associated inward calcium current. Another mode of action of NO/cGMP/PKGI signaling involves PKGI-mediated inactivation of the Ras homolog gene family member A (RhoA). Phosphorylation of RHOA by PRKG1 blocks the action of this protein in myriad processes: regulation of RHOA translocation; decreasing contraction; controlling vesicle trafficking, reduction of myosin light chain phosphorylation resulting in vasorelaxation. -

Supplemental Table 1A. Differential Gene Expression Profile of Adehcd40l and Adehnull Treated Cells Vs Untreated Cells
Supplemental Table 1a. Differential Gene Expression Profile of AdEHCD40L and AdEHNull treated cells vs Untreated Cells Fold change Regulation Fold change Regulation ([AdEHCD40L] vs ([AdEHCD40L] ([AdEHNull] vs ([AdEHNull] vs Probe Set ID [Untreated]) vs [Untreated]) [Untreated]) [Untreated]) Gene Symbol Gene Title RefSeq Transcript ID NM_001039468 /// NM_001039469 /// NM_004954 /// 203942_s_at 2.02 down 1.00 down MARK2 MAP/microtubule affinity-regulating kinase 2 NM_017490 217985_s_at 2.09 down 1.00 down BAZ1A fibroblastbromodomain growth adjacent factor receptorto zinc finger 2 (bacteria-expressed domain, 1A kinase, keratinocyte NM_013448 /// NM_182648 growth factor receptor, craniofacial dysostosis 1, Crouzon syndrome, Pfeiffer 203638_s_at 2.10 down 1.01 down FGFR2 syndrome, Jackson-Weiss syndrome) NM_000141 /// NM_022970 1570445_a_at 2.07 down 1.01 down LOC643201 hypothetical protein LOC643201 XM_001716444 /// XM_001717933 /// XM_932161 231763_at 3.05 down 1.02 down POLR3A polymerase (RNA) III (DNA directed) polypeptide A, 155kDa NM_007055 1555368_x_at 2.08 down 1.04 down ZNF479 zinc finger protein 479 NM_033273 /// XM_001714591 /// XM_001719979 241627_x_at 2.15 down 1.05 down FLJ10357 hypothetical protein FLJ10357 NM_018071 223208_at 2.17 down 1.06 down KCTD10 potassium channel tetramerisation domain containing 10 NM_031954 219923_at 2.09 down 1.07 down TRIM45 tripartite motif-containing 45 NM_025188 242772_x_at 2.03 down 1.07 down Transcribed locus 233019_at 2.19 down 1.08 down CNOT7 CCR4-NOT transcription complex, subunit 7 NM_013354