FRDC Final Report Design Standard
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Jolanta KEMPTER*, Maciej KIEŁPIŃSKI, Remigiusz PANICZ, and Sławomir KESZKA
ACTA ICHTHYOLOGICA ET PISCATORIA (2016) 46 (4): 287–291 DOI: 10.3750/AIP2016.46.4.02 MICROSATELLITE DNA-BASED GENETIC TRACEABILITY OF TWO POPULATIONS OF SPLENDID ALFONSINO, BERYX SPLENDENS (ACTINOPTERYGII: BERYCIFORMES: BERYCIDAE)—PROJECT CELFISH—PART 2 Jolanta KEMPTER*, Maciej KIEŁPIŃSKI, Remigiusz PANICZ, and Sławomir KESZKA Division of Aquaculture, West Pomeranian University of Technology, Szczecin, Kazimierza Krolewicza 4, 71-550 Szczecin, Poland Kempter J., Kiełpinski M., Panicz R., Keszka S. 2016. Microsatellite DNA-based genetic traceability of two populations of splendid alfonsino, Beryx splendens (Actinopterygii: Beryciformes: Berycidae)— Project CELFISH—Part 2. Acta Ichthyol. Piscat. 46 (4): 287–291. Background. The study is a contribution to Project CELFISH which involves genetic identifi cation of populations of fi sh species presenting a particular economic importance or having a potential to be used in the so-called commercial substitutions. The EU fi sh trade has been showing a distinct trend of more and more fi sh species previously unknown to consumers being placed on the market. Molecular assays have become the only way with which to verify the reliability of exporters. This paper is aimed at pinpointing genetic markers with which to label and differentiate between two populations of splendid alfonsino, Beryx splendens Lowe, 1834, a species highly attractive to consumers in Asia and Oceania due to the meat taste and low fat content. Material and methods. DNA was isolated from fragments of fi ns collected at local markets in Japan (MJ) (n = 10) and New Zealand (MNZ) (n = 18). The rhodopsin gene (RH1) fragment and 16 microsatellite DNA fragments (SSR) were analysed in all the individuals. -

A Preliminary Global Assessment of the Status of Exploited Marine Fish and Invertebrate Populations
A PRELIMINARY GLOBAL ASSESSMENT OF THE STATUS OF EXPLOITED MARINE FISH AND INVERTEBRATE POPULATIONS June 30 2018 A PRELIMINARY GLOBAL ASSESSMENT OF THE STATUS OF EXPLOITED MARINE FISH AND INVERTEBRATE POPULATIONS Maria. L.D. Palomares, Rainer Froese, Brittany Derrick, Simon-Luc Nöel, Gordon Tsui Jessika Woroniak Daniel Pauly A report prepared by the Sea Around Us for OCEANA June 30, 2018 A PRELIMINARY GLOBAL ASSESSMENT OF THE STATUS OF EXPLOITED MARINE FISH AND INVERTEBRATE POPULATIONS Maria L.D. Palomares1, Rainer Froese2, Brittany Derrick1, Simon-Luc Nöel1, Gordon Tsui1, Jessika Woroniak1 and Daniel Pauly1 CITE AS: Palomares MLD, Froese R, Derrick B, Nöel S-L, Tsui G, Woroniak J, Pauly D (2018) A preliminary global assessment of the status of exploited marine fish and invertebrate populations. A report prepared by the Sea Around Us for OCEANA. The University of British Columbia, Vancouver, p. 64. 1 Sea Around Us, Institute for the Oceans and Fisheries, University of British Columbia, 2202 Main Mall, Vancouver BC V6T1Z4 Canada 2 Helmholtz Centre for Ocean Research GEOMAR, Düsternbrooker Weg 20, 24105 Kiel, Germany TABLE OF CONTENTS Executive Summary 1 Introduction 2 Material and Methods 3 − Reconstructed catches vs official catches 3 − Marine Ecoregions vs EEZs 3 − The CMSY method 5 Results and Discussion 7 − Stock summaries reports 9 − Problematic stocks and sources of bias 14 − Stocks in the countries where OCEANA operates 22 − Stock assessments on the Sea Around Us website 31 − The next steps 32 Acknowledgements 33 References 34 Appendices I. List of marine ecoregions by EEZ 37 II. Summaries of number of stock by region and 49 by continent III. -

Elephant Fish
Best Fish Guide 2009-2010 How sustainable is New Zealand seafood? (Ecological Assessments) Produced and Published by Royal Forest and Bird Protection Society of New Zealand, Inc. PO Box 631, Level One, 90 Ghuznee Street, Wellington. www.forestandbird.org.nz November 2009 Acknowledgements Forest & Bird with to thank anonymous reviewers for their peer review comments on this draft. We also thank Peta Methias, Annabel Langbein, Martin Bosely, Margaret Brooker, Lois Daish, Kelder Haines, Dobie Blaze, Rohan Horner and Ray McVinnie for permission to use their recipes on the website. Special thanks to our Best Fish Guide Ambassador Dobie Blaze, keyboard player with Fat Freddy’s Drop. Editing: Kirstie Knowles, Barry Weeber and Helen Bain Technical Advisor: Barry Weeber Cover Design: Rob Deliver Cover fish (Tarakihi): Malcolm Francis Photography: Malcolm Francis: blue cod, blue moki, blue shark, butterfish, groper/hapuku, hoki, jack mackerel, john dory, kahawai, kingfish, leather jacket, moonfish, paua, porbeagle shark, red gurnard, red snapper, scallop, school shark, sea perch, snapper, spiny dogfish, tarakihi, trevally and trumpeter. Peter Langlands: blue warehou, cockles, elephantfish, frostfish, lookdown dory, oyster, pale ghost shark, queen scallops, red cod, rig/lemonfish, rubyfish and scampi. Ministry of Fisheries: albacore tuna, bigeye tuna, blue mackerel, pacific bluefin tuna, skipjack tuna, southern bluefin tuna and swordfish. John Holdsworth: gemfish, striped marlin and yellowfin tuna. Kirstie Knowles: sand flounder and rock lobster. Department of Conservation: kina and skate. Quentin Bennett: mako shark. Scott Macindoe: garfish. Jim Mikoz: yellow-eyed mullet. Forest & Bird: arrow squid, dark ghost shark, orange roughy, smooth oreo, packhorse lobster, paddle crabs, stargazer and white warehou. -

Fao Species Catalogue
FAO Fisheries Synopsis No. 125, Volume 15 ISSN 0014-5602 FIR/S125 Vol. 15 FAO SPECIES CATALOGUE VOL. 15. SNAKE MACKERELS AND CUTLASSFISHES OF THE WORLD (FAMILIES GEMPYLIDAE AND TRICHIURIDAE) AN ANNOTATED AND ILLUSTRATED CATALOGUE OF THE SNAKE MACKERELS, SNOEKS, ESCOLARS, GEMFISHES, SACKFISHES, DOMINE, OILFISH, CUTLASSFISHES, SCABBARDFISHES, HAIRTAILS AND FROSTFISHES KNOWN TO DATE FOOD AND AGRICULTURE ORGANIZATION OF THE UNITED NATIONS FAO Fisheries Synopsis No. 125, Volume 15 FIR/S125 Vol. 15 FAO SPECIES CATALOGUE VOL. 15. SNAKE MACKERELS AND CUTLASSFISHES OF THE WORLD (Families Gempylidae and Trichiuridae) An Annotated and Illustrated Catalogue of the Snake Mackerels, Snoeks, Escolars, Gemfishes, Sackfishes, Domine, Oilfish, Cutlassfishes, Scabbardfishes, Hairtails, and Frostfishes Known to Date I. Nakamura Fisheries Research Station Kyoto University Maizuru, Kyoto, 625, Japan and N. V. Parin P.P. Shirshov Institute of Oceanology Academy of Sciences Krasikova 23 Moscow 117218, Russian Federation FOOD AND AGRICULTURE ORGANIZATION OF THE UNITED NATIONS Rome, 1993 The designations employed and the presenta- tion of material in this publication do not imply the expression of any opinion whatsoever on the part of the Food and Agriculture Organization of the United Nations concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. M-40 ISBN 92-5-103124-X All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying or otherwise, without the prior permission of the copyright owner. Applications for such permission, with a statement of the purpose and extent of the reproduction, should be addressed to the Director, Publications Division, Food and Agriculture Organization of the United Nations, Via delle Terme di Caracalla, 00100 Rome, Italy. -

ILLEGAL FISHING Which Fish Species Are at Highest Risk from Illegal and Unreported Fishing?
ILLEGAL FISHING Which fish species are at highest risk from illegal and unreported fishing? October 2015 CONTENTS EXECUTIVE SUMMARY 3 INTRODUCTION 4 METHODOLOGY 5 OVERALL FINDINGS 9 NOTES ON ESTIMATES OF IUU FISHING 13 Tunas 13 Sharks 14 The Mediterranean 14 US Imports 15 CONCLUSION 16 CITATIONS 17 OCEAN BASIN PROFILES APPENDIX 1: IUU Estimates for Species Groups and Ocean Regions APPENDIX 2: Estimates of IUU Risk for FAO Assessed Stocks APPENDIX 3: FAO Ocean Area Boundary Descriptions APPENDIX 4: 2014 U.S. Edible Imports of Wild-Caught Products APPENDIX 5: Overexploited Stocks Categorized as High Risk – U.S. Imported Products Possibly Derived from Stocks EXECUTIVE SUMMARY New analysis by World Wildlife Fund (WWF) finds that over 85 percent of global fish stocks can be considered at significant risk of Illegal, Unreported, and Unregulated (IUU) fishing. This evaluation is based on the most recent comprehensive estimates of IUU fishing and includes the worlds’ major commercial stocks or species groups, such as all those that are regularly assessed by the United Nations Food and Agriculture Organization (FAO). Based on WWF’s findings, the majority of the stocks, 54 percent, are categorized as at high risk of IUU, with an additional 32 perent judged to be at moderate risk. Of the 567 stocks that were assessed, the findings show that 485 stocks fall into these two categories. More than half of the world’s most overexploited stocks are at the highest risk of IUU fishing. Examining IUU risk by location, the WWF analysis shows that in more than one-third of the world’s ocean basins as designated by the FAO, all of these stocks were at high or moderate risk of IUU fishing. -

Intrinsic Vulnerability in the Global Fish Catch
The following appendix accompanies the article Intrinsic vulnerability in the global fish catch William W. L. Cheung1,*, Reg Watson1, Telmo Morato1,2, Tony J. Pitcher1, Daniel Pauly1 1Fisheries Centre, The University of British Columbia, Aquatic Ecosystems Research Laboratory (AERL), 2202 Main Mall, Vancouver, British Columbia V6T 1Z4, Canada 2Departamento de Oceanografia e Pescas, Universidade dos Açores, 9901-862 Horta, Portugal *Email: [email protected] Marine Ecology Progress Series 333:1–12 (2007) Appendix 1. Intrinsic vulnerability index of fish taxa represented in the global catch, based on the Sea Around Us database (www.seaaroundus.org) Taxonomic Intrinsic level Taxon Common name vulnerability Family Pristidae Sawfishes 88 Squatinidae Angel sharks 80 Anarhichadidae Wolffishes 78 Carcharhinidae Requiem sharks 77 Sphyrnidae Hammerhead, bonnethead, scoophead shark 77 Macrouridae Grenadiers or rattails 75 Rajidae Skates 72 Alepocephalidae Slickheads 71 Lophiidae Goosefishes 70 Torpedinidae Electric rays 68 Belonidae Needlefishes 67 Emmelichthyidae Rovers 66 Nototheniidae Cod icefishes 65 Ophidiidae Cusk-eels 65 Trachichthyidae Slimeheads 64 Channichthyidae Crocodile icefishes 63 Myliobatidae Eagle and manta rays 63 Squalidae Dogfish sharks 62 Congridae Conger and garden eels 60 Serranidae Sea basses: groupers and fairy basslets 60 Exocoetidae Flyingfishes 59 Malacanthidae Tilefishes 58 Scorpaenidae Scorpionfishes or rockfishes 58 Polynemidae Threadfins 56 Triakidae Houndsharks 56 Istiophoridae Billfishes 55 Petromyzontidae -

Gemfish (Rexea Solandri) (Eastern Australian Population)
Advice to the Minister for the Environment, Heritage and the Arts from the Threatened Species Scientific Committee (the Committee) on Amendments to the list of Threatened Species under the Environment Protection and Biodiversity Conservation Act 1999 (EPBC Act) 1. Scientific name (common name) Rexea solandri (eastern Australian population) (Gemfish). This species is also known as Eastern Gemfish, Silver Gemfish and King Couta. 2. Reason for Conservation Assessment by the Committee This advice follows assessment of information provided by a public nomination to list Eastern Gemfish. This is the Committee’s first consideration of the species under the EPBC Act. 3. Summary of Conclusion The Committee judges that the eastern population of the species has been demonstrated to have met sufficient elements of Criterion 1 to make it eligible for listing as endangered, and has also been demonstrated to have met the requirements of section 179(6)(b) of the EPBC Act to be eligible for listing as conservation dependent. The Committee judges that the most appropriate category of listing for Eastern Gemfish is conservation dependent. 4. Taxonomy The species is conventionally accepted as Rexea solandri (Cuvier, 1831). 5. Description Gemfish are long, slender, silvery fish from the Family Gempylidae. They are similar in form to mackerel. Gemfish have a long, wide mouth with a protruding lower jaw and large fang- like teeth at the front. The species has a forked lateral line, with one branch running along the upper sides of the body, with the second branch diverging downward below the fifth dorsal fin spine, then running along the side of the body. -
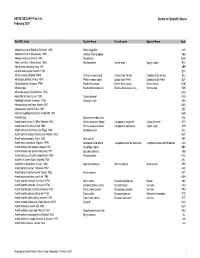
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -
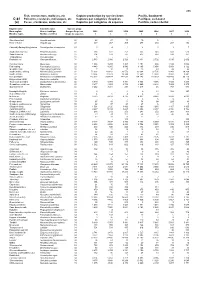
Fish, Crustaceans, Molluscs, Etc Capture Production by Species
495 Fish, crustaceans, molluscs, etc Capture production by species items Pacific, Southwest C-81 Poissons, crustacés, mollusques, etc Captures par catégories d'espèces Pacifique, sud-ouest (a) Peces, crustáceos, moluscos, etc Capturas por categorías de especies Pacífico, sudoccidental English name Scientific name Species group Nom anglais Nom scientifique Groupe d'espèces 2002 2003 2004 2005 2006 2007 2008 Nombre inglés Nombre científico Grupo de especies t t t t t t t Short-finned eel Anguilla australis 22 28 27 13 10 5 ... ... River eels nei Anguilla spp 22 337 267 209 277 210 207 152 Chinook(=Spring=King)salmon Oncorhynchus tshawytscha 23 0 4 1 2 1 1 7 Southern lemon sole Pelotretis flavilatus 31 238 322 251 335 348 608 513 Sand flounders Rhombosolea spp 31 204 193 187 437 514 530 351 Tonguefishes Cynoglossidae 31 3 - - - - - - Flatfishes nei Pleuronectiformes 31 2 580 2 986 2 729 3 431 2 702 3 015 2 602 Common mora Mora moro 32 1 308 1 234 1 403 1 154 986 1 180 1 088 Red codling Pseudophycis bachus 32 4 443 8 265 9 540 8 165 5 854 5 854 6 122 Grenadier cod Tripterophycis gilchristi 32 7 10 13 13 43 29 26 Southern blue whiting Micromesistius australis 32 72 203 43 812 26 576 30 304 32 735 23 943 29 268 Southern hake Merluccius australis 32 13 834 22 623 19 344 12 560 12 858 13 892 8 881 Blue grenadier Macruronus novaezelandiae 32 215 302 209 414 147 032 134 145 119 329 103 489 96 119 Ridge scaled rattail Macrourus carinatus 32 - - - - - 9 14 Thorntooth grenadier Lepidorhynchus denticulatus 32 5 349 5 304 6 341 3 855 4 056 3 725 3 264 Grenadiers, rattails nei Macrouridae 32 3 877 4 253 3 732 2 660 2 848 7 939 8 970 Gadiformes nei Gadiformes 32 3 252 3 281 298 1 217 46 767 886 Broadgilled hagfish Eptatretus cirrhatus 33 2 - 0 0 11 508 347 Sea catfishes nei Ariidae 33 4 6 4 4 4 .. -

Supplementary Information Doi: 10.1038/Nclimate1691
SUPPLEMENTARY INFORMATION DOI: 10.1038/NCLIMATE1691 Shrinking of fishes exacerbates impacts of global ocean changes on marine ecosystems Supplementary information Method Dynamic Bioclimate Envelope Model (DBEM) As a first step, we predicted the current (19702000) distribution map of each species an algorithm described in [1,2]. This algorithm estimates the relative abundance of a species on a 30’ latitude x 30’ longitude grid of the world ocean. Input parameters for DBEM include the species’ maximum and minimum depth limits, northern and southern latitudinal range limits, an index of association to major habitat types (seamounts, estuaries, inshore, offshore, continental shelf, continental slope and the abyssal) and known occurrence boundaries. The parameter values of each species, which are posted on the Sea Around Us Project website (http://www.seaaroundus.org/distribution/search.aspx), were derived from data in online databases, mainly FishBase (www.fishbase.org). Jones et al. 3 compared the predicted species distribution from this algorithm with empirically observed occurrence records and found that the algorithm has high predictive power, and that its skills are comparable with other commonly used species distribution modelling approach for marine species such as Maxent4 and Aquamap5. As a second step, we used DBEM to identify the ‘environmental preference profiles’ of the studied species, defined by outputs from the Earth System Models, including sea water temperature (bottom and surface), depth, salinity, distance from seaice and habitat types. Preference profiles are defined as the suitability of each of these environmental conditions to each species, with suitability calculated by overlaying environmental data (19702000) with maps of relative abundance of the species 6. -
Docket No. 201029-0282]
This document is scheduled to be published in the Federal Register on 11/09/2020 and available online at federalregister.gov/d/2020-24416, and on govinfo.gov BILLING CODE 3510-22-P DEPARTMENT OF COMMERCE National Oceanic and Atmospheric Administration 50 CFR Part 216 [Docket No. 201029-0282] RIN 0648-XG809 Implementation of Fish and Fish Product Import Provisions of the Marine Mammal Protection Act--Notification of Rejection of Petition and Issuance of Comparability Findings AGENCY: National Marine Fisheries Service (NMFS), National Oceanic and Atmospheric Administration (NOAA), Commerce. ACTION: Denial of petition and issuance of comparability findings. SUMMARY: Under the authority of the Marine Mammal Protection Act (MMPA), the NMFS Assistant Administrator for Fisheries (Assistant Administrator) has denied a petition for emergency rulemaking from Sea Shepherd Legal. Additionally, the Assistant Administrator has issued comparability findings for the Government of New Zealand’s (GNZ) following fisheries: West Coast North Island multi-species set net fishery, and West Coast North Island multi-species trawl fishery. NMFS bases the comparability findings on documentary evidence submitted by the GNZ and other relevant, readily-available information including the scientific literature. DATES: These comparability findings are valid for the period of [INSERT DATE OF FILING FOR PUBLIC INSPECTION WITH THE OFFICE OF THE FEDERAL REGISTER], through January 1, 2023, unless revoked by the Assistant Administrator in a subsequent action. FOR FURTHER -

The Sea & Freshwater Fishes of Australia & New Guinea
Calypso Publications C. 13 1998 The Sea & Freshwater Fishes of Australia & New Guinea Part One South-East , South , South-West Australia & Tasmania (in part) The 1997/8 Classified Taxonomic Checklist Also Available in the Calypso Taxonomic Series :- The Sea & Freshwater Fishes of Australia & New A Classified Taxonomic Checklist of all verified & Guinea. validated species recorded on the Calypso Ichthyological Database of Marine and Part Two Freshwater Fishes from Southern Australia and Australia .North-West /North /North East & New Guinea Tasmania (in part) Calypso Database Areas 08- & 98-. ISBN :0906301 60 2 Gerald Jennings A Word from Calypso... The taxonomic information on the enclosed disk has been extracted from the Calypso Ichthyological Database of the World's Fishes . An introductory booklet to this system is also published as a separate document (ISBN 0906301 47 5) Information on the main database is necessarily stored on several different smaller databases and some of the material on these databases has not yet been uniformly organised. Although this fact in no way affects the validity of the information provided here, an explanation of these minor anomalies was felt necessary. Database Identification Numbers invariably consist of six numerals in a three + three format, and each number is unique to a particular Species - even if the species name is changed at a later date the number remains valid. The species numbers can convey additional limited information in themselves and a sheet giving a breakdown of this information is also enclosed. Later disks may include this information under 'help'. Some numbers in some contexts are suffixed by a letter but this letter will probably only be of use in the context of written publications.