Exploring Genetic Interactions and Networks with Yeast
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Evolution of Genetic Systems in Filamentous Ascomycetes
Evolution of Genetic Systems in Filamentous Ascomycetes Evolutie van genetische systemen in hyphenvormende zakjeszwammen 0000 0513 3836 Promotor: dr. R.F. Hoekstra hoogleraar in de populatie- en kwantitatieve genetica fjtfoiißi f ßin Maarten J. Nauta Evolution of Genetic Systems in Filamentous Ascomycetes Proefschrift ter verkrijging van de graad van doctor in de landbouw- en milieuwetenschappen op gezag van de rector magnificus, dr. C.M. Karssen, in het openbaar te verdedigen op woensdag 12januar i 1994 des namiddags te vier uur in de Aula van de Landbouwuniversiteit te Wageningen. 15 0 S(p^ZJ> These investigations were supported by the Netherlands Organization for Scientific Research (N.W.O.). BibUt/FHEEK LAMDbOirWUNIVERSITEJi. WAGE NINGE N CIP-GEGEVENS KONINKLIJKE BIBLIOTHEEK, DEN HAAG Nauta, Maarten J. Evolution of genetic systems in filamentous ascomycetes / Maarten J. Nauta. - [ S.l. : s.n.]. -111 . Thesis Wageningen. - With ref. - With summary in Dutch. ISBN 90-5485-199-6 Subject headings: population genetics / ascomycetes. omslagontwerp: Ernst van Cleef foto omslag: Barrages tussen verschillende stammen van Podospora anserina als gevolg van vegetatieve incompatibiliteit. (met dank aan Inge Haspels) aan mijn ouders Voorwoord Dit proefschrift is het resultaat van vier jaar onderzoek, verricht bij de vakgroep Erfelijkheidsleer van de Landbouwuniversiteit in Wageningen. In zekere zin valt zo'n proefschrift te vergelijken met een levend wezen. Uit de genetica is bekend dat de verschijningsvorm van elk levend wezen tot stand komt door een combinatie van erfelijke aanleg en invloeden uit de omgeving. Voor een proefschrift geldt eigenlijk hetzelfde: Zowel het werk van de auteur, als de bijdragen van zijn omgeving zijn onontbeerlijk om tot een verschijningsvorm te komen. -

Self-Fertility and Uni-Directional Mating-Type Switching in Ceratocystis Coerulescens, a Filamentous Ascomycete
Curr Genet (1997) 32: 52–59 © Springer-Verlag 1997 ORIGINAL PAPER T. C. Harrington · D. L. McNew Self-fertility and uni-directional mating-type switching in Ceratocystis coerulescens, a filamentous ascomycete Received: 6 July 1996 / 25 March 1997 Abstract Individual perithecia from selfings of most some filamentous ascomycetes. Although a switch in the Ceratocystis species produce both self-fertile and self- expression of mating-type is seen in these fungi, it is not sterile progeny, apparently due to uni-directional mating- clear if a physical movement of mating-type genes is in- type switching. In C. coerulescens, male-only mutants of volved. It is also not clear if the expressed mating-types otherwise hermaphroditic and self-fertile strains were self- of the respective self-fertile and self-sterile progeny are sterile and were used in crossings to demonstrate that this homologs of the mating-type genes in other strictly heter- species has two mating-types. Only MAT-2 strains are othallic species of ascomycetes. capable of selfing, and half of the progeny from a MAT-2 Sclerotinia trifoliorum and Chromocrea spinulosa show selfing are MAT-1. Male-only, MAT-2 mutants are self- a 1:1 segregation of self-fertile and self-sterile progeny in sterile and cross only with MAT-1 strains. Similarly, self- perithecia from selfings or crosses (Mathieson 1952; Uhm fertile strains generally cross with only MAT-1 strains. and Fujii 1983a, b). In tetrad analyses of selfings or crosses, MAT-1 strains only cross with MAT-2 strains and never self. half of the ascospores in an ascus are large and give rise to It is hypothesized that the switch in mating-type during self-fertile colonies, and the other ascospores are small and selfing is associated with a deletion of the MAT-2 gene. -

MEIOSIS and RECOMBINATION in SORDARIA FIMICOLA Introduction
MEIOSIS AND RECOMBINATION IN SORDARIA FIMICOLA Introduction: In ascomycete fungi, a form of meiosis occurs in which the products of meiosis order themselves within a fruiting body according to the physical separation and segregation of chromatids during the meiotic process. This is covered in some detail on pages 150-152 (including Figures 4.26 and 4.27) in Hartl and Jones, Essential Genetics. You should study these pages before beginning this module. As described, ordered tetrad analysis provides a way to measure the genetic map distance between a gene and the centromere of the chromosome on which that gene resides. That is what you will do over the next two weeks in this laboratory. I. Natural history and Life Cycle of Sordaria fimicola Sordaria fimicola is an ascomycete fungi that can be found growing in rotting vegetation and animal dung (in fact, the name Sordaria fimicola means "filthy dung dweller"). Sordaria and another ascomycete, the common bread fungus Neurospora crassa (Fig. 4.26), have been used as model systems for studying the process of chromosome exchange (crossing-over) because of their reproductive characteristics. The life cycle of Sordaria is representative of the ascomycetes (although there are substantial differences in the details among species). The individual fungus begins as a haploid ascospore. The ascospore germinates to form hyphae (singular = hypha), which are long filaments comprised of haploid cells. These hyphae grow and extend throughout the nutrient source (dung or rotting vegetation in nature, nutrient medium in the laboratory situation) and digest it by means of enzymes secreted by the cells. Nutrients are then absorbed into the cells. -

Advanced Gene Mapping in Eukaryotes
RUSSMC07 2/7/05 11:54 AM Page 165 7 Advanced Gene Mapping in Eukaryotes Ordered tetrads of the fungus, Neurospora Crassa. PRINCIPAL POINTS • Tetrad analysis is a mapping technique that can be be used to determine gene order and map distances used to map the genes of certain haploid eukaryotic between genes. organisms in which the products of a single meiosis— • Due to both ethical and practical issues, human the meiotic tetrad—are contained within a single genes cannot be mapped by making crosses and structure. In these situations, the map distance analyzing progeny. A number of approaches are between genes is computed by analyzing the relative used to determine the linkage relationships proportion of tetrad types, rather than by analyzing between human genes, including analyzing individual progeny. pedigree data recombinationally and physically • In organisms in which the meiotic tetrads are locating genes on chromosomes by molecularly arranged linearly, it is easy to map the distance aided methods. of a gene from its centromere. • Crossing-over can also occur during mitosis, although i SPECIAL TECHNIQUES ARE AVAILABLE FOR at a frequency much lower than during meiosis. As in constructing linkage maps for eukaryotic organisms meiosis, mitotic crossing-over occurs at a four-strand beyond the standard genetic mapping crosses. In this stage. chapter you will learn about these techniques. Then, in the iActivity, you can apply what you’ve learned and further • For organisms amenable to such analysis, such as the explore one of the advanced mapping techniques. fungus Aspergillus nidulans, mitotic recombination can iActivity 165 RUSSMC07 1/26/05 12:47 PM Page 166 166 Chapter 7 Advanced Gene Mapping in Eukaryotes In the previous chapter, we considered the classical princi- Go to the iActivity Mapping Genes by Tetrad i ples for mapping genes in eukaryotes by means of recombi- Analysis on your CD-ROM and assume the role nation analysis. -
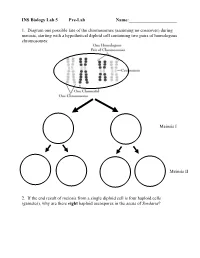
INS Biology Lab 5 Pre-Lab Name:______
INS Biology Lab 5 Pre-Lab Name:_____________________ 1. Diagram one possible fate of the chromosomes (assuming no crossover) during meiosis, starting with a hypothetical diploid cell containing two pairs of homologous chromosomes: Meiosis I Meiosis II 2. If the end result of meiosis from a single diploid cell is four haploid cells (gametes), why are there eight haploid ascospores in the ascus of Sordaria? 2 Winter Lab 1. Meiosis This lab exercise courtesy of Mike Clayton, University of Wisconsin Adapted from a lab written by Jon Glase, Cornell University I. The Process of Meiosis Meiosis is a process of two sequenced nuclear divisions neither of which is identical to mitosis. Meiosis always begins with a nucleus with two sets of chromosomes (a diploid nucleus) where each chromosome has two chromatids. Meiosis produces four nuclei each with one set of chromosomes (haploid nuclei) in which each chromosome has one chromatid. Meiosis together with syngamy constitutes sexuality in eukaryotes. Sexual reproduction generates individuals with new and unique genotypic combinations each generation. Meiosis, by itself, can generate new combinations of genetic material. It does so in two ways. -- through the independent assortment of different homologous pairs of chromosomes and -- through crossing over between homologous pairs of chromosomes. Pop Beads as Models of Chromosomes The shuffling and recombination of genetic material can be modeled using pop beads. Before lab, preview this web page to see a demonstration of meiosis using pop beads ( http://botit.botany.wisc.edu/courses/botany_130/Meiosis/pop.html ). In lab, you will work with a partner to recreate the following scenarios of meiosis.. -

Chapter 1 a Study of Gene Linkage and Mapping Using Tetrad Analysis
Chapter 1 A Study of Gene Linkage and Mapping Using Tetrad Analysis in the Fungus Sordaria fimicola Jon C. Glase Introductory Biology Program Division of Biological Sciences Cornell University Ithaca, New York 14853-0901 (607) 255-3007, FAX (607) 255-1301, [email protected] Jon is a Senior Lecturer in the Introductory Biology Program at Cornell University, where he is coordinator of the introductory biology laboratory course for biology majors. He received his B.S. in Biology (1967) and Ph. D. in Behavioral Ecology (1971) from Cornell. He was a co-founder of ABLE and President from 1991–1993. His research interests include social organization of bird flocks, laboratory curriculum development, and computer biology simulations. Reprinted from: Glase, J. C. 1995. A study of gene linkage and mapping using tetrad analysis in the fungus Sordaria fimicola. Pages 1–24, in Tested studies for laboratory teaching, Volume 16 (C. A. Goldman, Editor). Proceedings of the 16th Workshop/Conference of the Association for Biology Laboratory Education (ABLE), 273 pages. Although the laboratory exercises in ABLE proceedings volumes have been tested and due consideration has been given to safety, individuals performing these exercises must assume all responsibility for risk. The Association for Biology Laboratory Education (ABLE) disclaims any liability with regards to safety in connection with the use of the exercises in its proceedings volumes. © 1995 Cornell University 1 2 Tetrad Analysis Contents Introduction ......................................................................................................2 -

Homothallism Expression in Kluyveromyces Lactis
Heredity (1983), 51(3),549—559 1983. The Genetical Society of Great Britain HOMOTHALLISMEXPRESSION IN KLUYVEROMYCES LACTIS ë. NOVOTN, J.-L FUENTES, P. CARRE, H. BOZE, G. MOULIN, AND P. GALZY Chairede Genefique et Microbiologie, ENSA, 34060 Montpellier Cedex, France Received2.iii.83 SUMMARY Kinetics of the mating-type interconversion was observed after heterothallic nutritionally-marked stocks of Kluyveromyces lactis were mated and sporulated. Conversion to homothallism is not finished within the period of 10 days of growth on the sporulation agar. In general, the expression of homothallism in the a mating-type is often delayed when compared to the a mating-type. Segregation patterns of tetrads suggest the functioning of a mating-type intercon- version system including a mating locus and two silent storage copies of the mating-type information. 1. INTRODUCTION Homothallism of yeast has lately attracted much attention. It endows monoclonal cultures of yeast with a striking power to diploidise and sub- sequently to sporulate. Some scientists believe that it also serves as an example of a simple differentiating system from which conclusions may be drawn about the general mechanisms controlling differentiation in multi- cellular eukaryotic organisms. The mating-type locus MAT of Saccharomyces cerevisiae exists in two states, a or a, which control the ability of yeast cells to mate and sporulate (Mortimer and Hawthorne, 1969). Two genes HMLa and HMRa were found to be necessary for the conversion of mating-types, and shown to map on chromosome III, loosely linked with the MAT locus (Harashima and Oshima, 1976). Observations that both a HO hmra hmla and a HO hmra hmla genotypes containing only "inactive" alleles of HMLa and HMRa had been able to switch mating-types, lead to a conclusion that the "inactive" alleles hmra and hmla were equivalent to HMLa and HMRa, respectively (Naumov and Tolstrukov, 1973; Harashima et aL, 1974; Harashima and Oshima, 1976). -

Aeiab Meiosis
Topic 11. Meiosis I. The Process of Meiosis Meiosis is a process of two sequenced nuclear divisions neither of which is identical to mitosis. Meiosis always begins with a nucleus with two sets of chromosomes (a diploid nucleus) where each chromosome has two chromatids. Meiosis produces four nuclei each with one set of chromosomes (haploid nuclei) in which each chromosome has one chromatid. Meiosis together with syngamy constitutes sexuality in eukaryotes. Sexual reproduction generates individuals with new and unique genotypic combinations each generation. Meiosis, by itself, can generate new combinations of genetic material. It does so in two ways. - through the independent assortment of different homologous pairs of chromosomes and - through crossing over between homologous pairs of chromosomes. Pop Beads as Models of Chromosomes The shuffling and recombination of genetic material can be modeled using pop beads. Your TA will illustrate the process of meiosis for you using pop beads...... 1. with one pair of homologs without consideration of crossing over to simply demonstrate the differences between meiosis and mitosis. 2. with two pairs of homologs without consideration of crossing to demonstrate the independent movement of differently pairs of homologs. 3. with one pair of homologs showing crossing over to demonstrate how the genetic information is exchanged between homologous chromosomes. You should then work with a partner to recreate these scenarios, and to do a fourth with two sets of homologs and with crossing over. Note, we have a lesson on the web page that clearly explains how we use pop beads to model chromosomes in meiosis. You will need to be able to interpret questions about the process of meiosis that requires both an understanding of meiosis, and also of how pop beads are used in lab to represent chromosomes in the various stages of meiosis. -

Meiotic Recombination and Genomic Diversity in the Yeast Komagataella
bioRxiv preprint doi: https://doi.org/10.1101/704627; this version posted July 16, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 3 Tetrad analysis without tetrad dissection: Meiotic recombination and genomic 4 diversity in the yeast Komagataella phaffii (Pichia pastoris) 5 6 1 1,2 1 1 7 Stephanie Braun-Galleani , Julie A. Dias , Aisling Y. Coughlan , Adam P. Ryan , 1 1 8 Kevin P. Byrne , Kenneth H. Wolfe * 9 10 1 11 UCD Conway Institute, School of Medicine, University College Dublin, Dublin, Ireland, 2 12 Department of Mathematics and Statistics, McGill University, Montreal, Quebec, Canada. 13 14 * [email protected] (KHW) 15 16 Short title: Tetrad analysis in Komagataella phaffii 17 1 bioRxiv preprint doi: https://doi.org/10.1101/704627; this version posted July 16, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 18 Abstract 19 Komagataella phaffii is a yeast widely used in the pharmaceutical and biotechnology 20 industries, and is one of the two species that were formerly called Pichia pastoris. However, 21 almost all laboratory work on K. phaffii has been done on strains derived from a single 22 natural isolate, CBS7435. There is little information about the genetic properties of K. -

Some Genetic Interrelations of Methionine Mutants of Neurospora
SOME GENETIC INTERRELATIONS OF METHIONINE MUTANTS OF NEUROSPORA CRASSA A Thesis submitted by NOREEN ELIZABETH MURRAY a candidate for the degree of PHILOSOPHIAE DOCTOR University of Birmingham October, 1959 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ACKNOWLEDGEMENTS The author wishes to express her thanks to Professor D.&. Catcheside, F.R.S. for his encouragement and heipful criticism given throughout these investi gations, and to Dr. J.R.S. Fincham who kindly donated a number of methionine mutants. Grateful thanks are also due to the Department of Scientific and Industrial Research for a maintenanc*' grant. SYW0PSI3 Methionine nutants of Neurosnora crassa v/ere isolated hy a filtration concentration technique fclloT!^ing nltra-Yiclet Ipradiation. These and other mutants were classified by the physiological tests of precursor utilisation and heterocaryon complementation. precursor utilisation data are in agreement with the present outline of methionine synthesis from cysteine but provide little infoimstion on the pathway of sulphate reduction and incorporation of sulphur within a carbon skeleton to yield cysteine. Seven cysteine and eight methionine loci are represented on the basis of heterocaryon complementation tests. -

MARKERS and CENTROMERE DISTANCES in NEUROSPORA Tetraspermal
MARKERS AND CENTROMERE DISTANCES IN NEUROSPORA TETRASPERMAl H. BRANCH HOWE, JR. Department of Bacteriology, University of Georgia, Athens, Georgia Received September 24, 1962 ENETIC studies with Neurospora tetrasperma were begun by the late B. 0. GD~~~~( 1928) soon after the genus Neurospora was described (SHEARand DODGE1927). DODGE(1927) showed that the linear ascus of N. tetrasperm contains four binucleate ascospores and that events during ascus formation usually provide that the two nuclei in each ascospore will differ in mating type. The four ascospores from a single ascus, therefore, usually yield four self-fertile mycelia, a phenomenon variously known as facultative, secondary, or pseudo- homothallism. Genetic differences studied include mating type (DODGE1928; LINDEGREN 1932) ; conidial variation (DODGE1930,1935) ;lethals ( UBERand GODDARD1934; DODGE1934, 1935; SEAVER1937; DODGE,SINGLETON and ROLNICK1950); substrate color (TAI1936); dwarfism (DODGE,SCHMIDT and APPEL1945); and other morphological traits ( GOODSPEED1942; MALLOCH1942). Until recently (HOWE1961) mainly visible differences were employed. Genetic work in N. tetrasperma clearly lags behind that in N. crmsa both in numbers of mutants produced and in linkage analysis. Linkage analysis in N. tetrmperma is compli- cated by nuclear passing during ascus formation (DODGE1927) and by the di- caryotic condition of the ascospores. Single ascospore isolation does not neces- sarily insure homokaryosis for every allele, as in N.crassa. The present investigation involves the recovery of both morphological and nutritional mutants as well as a preliminary determination of gene-centromere distances using both tetrads and random ascospores. The genetic analyses are based upon current understanding of ascus formation and the relationship be- tween crossing over and nuclear content of the ascospores. -

"Budding Yeast Saccharomyces Cerevisiae As a Model Genetic
k Budding Yeast Introductory article Article Contents Saccharomyces cerevisiae • Introduction • How Yeasts Became Popular Model Systems as a Model Genetic • Yeast Biology • Research Milestones Organism • The Awesome Power of Yeast Genetics • Conclusion Sean M Burgess, University of California at Davis, Davis, California, USA • Acknowledgements University of California at Davis, Davis, California, USA Ted Powers, Online posting date: 15th November 2017 Joshua Chang Mell, Department of Microbiology and Immunology, Drexel Uni- versity College of Medicine, Philadelphia, Pennsylvania, USA Budding yeast has served as an experimental high-throughput analysis have allowed for the elucidation of organism for genetic research for over 50 years. the gene networks that underlie eukaryotic cellular function. The yeast shares a common cell division cycle Analysis of these networks has advanced our understanding of and cellular architecture with other eukaryotes, disease. Conservation of yeast genes extends to over 1000 human disease genes (Heinicke et al., 2007) including the genetic basis and as a microorganism, it is easily propagated and of birth defects, cancer and even neurodegenerative disorders manipulated in the laboratory. An intense focus such as Parkinson’s, Alzheimer’s and Huntington’s diseases. The on the central dogma, the cell cycle and sexual long history of yeast research has led to a wealth of tools and reproduction unleashed new fields including gene information, utilised not only by yeast researchers but also by silencing, homologous recombination and differ- the field of genetics as a whole. Collectively, these features con- ential gene expression, among others. The ease tribute to what yeast researchers fondly refer to as the ‘awesome k of genetic analysis allowed researchers to study power of yeast genetics’! See also: Yeast as a Model for Human k processes to a degree not seen for other model Diseases organisms.