Atomistic Simulation for Transition Metal Dichalcogenides Using NEMO5 and Medea-VASP
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Magnetic Field Enhanced Superconductivity in Epitaxial Thin Film Wte2
Lawrence Berkeley National Laboratory Recent Work Title Magnetic Field Enhanced Superconductivity in Epitaxial Thin Film WTe2. Permalink https://escholarship.org/uc/item/1642v6qf Journal Scientific reports, 8(1) ISSN 2045-2322 Authors Asaba, Tomoya Wang, Yongjie Li, Gang et al. Publication Date 2018-04-25 DOI 10.1038/s41598-018-24736-x Peer reviewed eScholarship.org Powered by the California Digital Library University of California www.nature.com/scientificreports OPEN Magnetic Field Enhanced Superconductivity in Epitaxial Thin Film WTe2 Received: 13 December 2017 Tomoya Asaba1, Yongjie Wang 2, Gang Li 1, Ziji Xiang1, Colin Tinsman1, Lu Chen1, Accepted: 5 April 2018 Shangnan Zhou1, Songrui Zhao2, David Laleyan2, Yi Li3, Zetian Mi2 & Lu Li 1 Published: xx xx xxxx In conventional superconductors an external magnetic feld generally suppresses superconductivity. This results from a simple thermodynamic competition of the superconducting and magnetic free energies. In this study, we report the unconventional features in the superconducting epitaxial thin flm tungsten telluride (WTe2). Measuring the electrical transport properties of Molecular Beam Epitaxy (MBE) grown WTe2 thin flms with a high precision rotation stage, we map the upper critical feld Hc2 at diferent temperatures T. We observe the superconducting transition temperature Tc is enhanced by in-plane magnetic felds. The upper critical feld Hc2 is observed to establish an unconventional non- monotonic dependence on temperature. We suggest that this unconventional feature is due to the lifting of inversion symmetry, which leads to the enhancement of Hc2 in Ising superconductors. Superconductivity generally competes with magnetic felds. Based on thermodynamics, an applied magnetic feld usually suppresses superconductivity by destroying the underlying electron pairing in the superconducting state1. -

Wte2): an Atomic Layered Semimetal
The Pennsylvania State University The Graduate School Department of Materials Science and Engineering TUNGSTEN DITELLURIDE (WTE2): AN ATOMIC LAYERED SEMIMETAL A Thesis in Materials Science and Engineering by Chia-Hui Lee 2015 Chia-Hui Lee Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science December 2015 ii The thesis of Chia-Hui Lee was reviewed and approved* by the following: Joshua A. Robinson Professor of Materials Science and Engineering Thesis Advisor Thomas E. Mallouk Head of the Chemistry Department Evan Pugh University Professor of Chemistry, Physics, Biochemistry and Molecular Biology Mauricio Terrones Professor of Physics, Chemistry and Materials Science & Engineering Suzanne Mohney Professor of Materials Science and Engineering and Electrical Engineering Chair, Intercollege Graduate Degree Program in Materials Science and Engineering *Signatures are on file in the Graduate School iii ABSTRACT Tungsten ditelluride (WTe2) is a transition metal dichalcogenide (TMD) with physical and electronic properties that make it attractive for a variety of electronic applications. Although WTe2 has been studied for decades, its structure and electronic properties have only recently been correctly described. We explored WTe2 synthesis via chemical vapor transport (CVT) method for bulk crystal, and chemical vapor deposition (CVD) routes for thin film material. We employed both experimental and theoretical techniques to investigate its structural, physical and electronic properties of WTe2, and verify that WTe2 has its minimum energy configuration in a distorted 1T structure (Td structure), which results in metallic-like behavior. Our findings confirmed the metallic nature of WTe2, introduce new information about the Raman modes of Td-WTe2, and demonstrate that Td- WTe2 is readily oxidized via environmental exposure. -

View PDF Version
Nanoscale Advances View Article Online REVIEW View Journal | View Issue In-plane anisotropic electronics based on low- symmetry 2D materials: progress and prospects Cite this: Nanoscale Adv., 2020, 2,109 Siwen Zhao, a Baojuan Dong,bc Huide Wang,a Hanwen Wang,bc Yupeng Zhang, a Zheng Vitto Han*bc and Han Zhang *a Low-symmetry layered materials such as black phosphorus (BP) have been revived recently due to their high intrinsic mobility and in-plane anisotropic properties, which can be used in anisotropic electronic and optoelectronic devices. Since the anisotropic properties have a close relationship with their anisotropic structural characters, especially for materials with low-symmetry, exploring new low- symmetry layered materials and investigating their anisotropic properties have inspired numerous research efforts. In this paper, we review the recent experimental progresses on low-symmetry layered Received 4th October 2019 materials and their corresponding anisotropic electrical transport, magneto-transport, optoelectronic, Accepted 30th October 2019 thermoelectric, ferroelectric, and piezoelectric properties. The boom of new low-symmetry layered DOI: 10.1039/c9na00623k materials with high anisotropy could open up considerable possibilities for next-generation anisotropic Creative Commons Attribution 3.0 Unported Licence. rsc.li/nanoscale-advances multifunctional electronic devices. 1. Introduction energy band structure and can be regarded as a process of lowering the dimensionality of the carrier transport. Therefore, Two dimensional (2D) layered materials with strong in-plane the electrical, optical, thermal, and phonon properties of these covalent bonds and weak out-of-plane van der Waals interac- anisotropic materials are diverse along the different in-plane tions span a very broad range of solids and exhibit extraordinary crystal directions. -
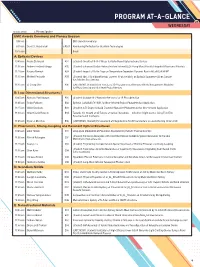
Program-At-A-Glance (PDF)
PROGRAM AT-A-GLANCE WEDNESDAY All times are EDT Plenary Speaker EMC Awards Ceremony and Plenary Session 9:00 am EMC Awards Ceremony 9:15 am David D. Awschalom PL01 Abandoning Perfection for Quantum Technologies 10:15 am Break A: Epitaxial Devices 10:45 am Rasha El-Jaroudi A01 (Student) Growth of B-III-V Alloys for GaAs-Based Optoelectronic Devices 11:00 am Andrew Frederick Briggs A02 (Student) Enhanced Double Heterostructure Infrared LEDs Using Monolithically Integrated Plasmonic Materials 11:15 am Nayana Remesh A03 (Student) Impact of Buffer Traps on Temperature-Dependent Dynamic Ron in AlGaN/GaN HEMT 11:30 am Michael Pedowitz A05 (Student) Mn+3 Rich Nanofiberous Layeredδ -phase MnO2 on Epitaxial Graphene-Silicon Carbide for Selective Gas Sensing 11:45 am Li-Chung Shih A06 (LATE NEWS, Student) Dual-Function ZTO Phototransistor Memory with Au Nanoparticles Mediated for Photo-Sensing and Multilevel Photo-Memory B: Low-Dimensional Structures I 10:45 am Nicholas Paul Morgan B01 (Student) Scalable III-V Nanowire Networks for IR Photodetection 11:00 am Rabin Pokharel B02 Epitaxial GaAsSbN (Te) NWs for Near-Infrared Region Photodetection Application 11:15 am Shisir Devakota B03 (Student) A Te Doped GaAsSb Ensemble Nanowire Photodetector for Near-Infrared Application 11:30 am Gilbert Daniel Nessim B04 Towards the Growth of 3D Forests of Carbon Nanotubes—Selective Height Control Using Thin-Film Reservoirs and Overlayers 11:45 am Dylan J. McIntyre B06 (LATE NEWS, Student) Enhancement of Integrated Cu-Ti-CNT Conductors via Joule-Heating Driven -

View PDF Version
Nanoscale View Article Online REVIEW View Journal | View Issue Synthesis of emerging 2D layered magnetic materials Cite this: Nanoscale, 2021, 13, 2157 Mauro Och,a Marie-Blandine Martin,b Bruno Dlubak, b Pierre Seneorb and Cecilia Mattevi *a van der Waals atomically thin magnetic materials have been recently discovered. They have attracted enormous attention as they present unique magnetic properties, holding potential to tailor spin-based device properties and enable next generation data storage and communication devices. To fully under- stand the magnetism in two-dimensions, the synthesis of 2D materials over large areas with precise thick- ness control has to be accomplished. Here, we review the recent advancements in the synthesis of these materials spanning from metal halides, transition metal dichalcogenides, metal phosphosulphides, to ternary metal tellurides. We initially discuss the emerging device concepts based on magnetic van der Waals materials including what has been achieved with graphene. We then review the state of the art of Creative Commons Attribution-NonCommercial 3.0 Unported Licence. the synthesis of these materials and we discuss the potential routes to achieve the synthesis of wafer- scale atomically thin magnetic materials. We discuss the synthetic achievements in relation to the struc- Received 3rd November 2020, tural characteristics of the materials and we scrutinise the physical properties of the precursors in relation Accepted 8th January 2021 to the synthesis conditions. We highlight the challenges related to the synthesis of 2D magnets and we DOI: 10.1039/d0nr07867k provide a perspective for possible advancement of available synthesis methods to respond to the need for rsc.li/nanoscale scalable production and high materials quality. -

Large-Area and High-Quality 2D Transition Metal Telluride
Large-area and high-quality 2D transition metal telluride Jiadong Zhou1†, Fucai Liu1†, Junhao Lin2,3,4*, Xiangwei Huang5†, Juan Xia6, Bowei Zhang1, Qingsheng Zeng1, Hong Wang1, Chao Zhu1, Lin Niu1, Xuewen Wang1, Wei Fu1, Peng Yu1, Tay- Rong Chang7, Chuang-Han Hsu8,9, Di Wu8,9, Horng-Tay Jeng7,10, Yizhong Huang1, Hsin Lin8,9, , Zexiang Shen1,6,11, Changli Yang5,12, Li Lu5,12, Kazu Suenaga4, Wu Zhou2, Sokrates T. Pantelides2,3, Guangtong Liu5* and Zheng Liu1, 13,14*. 1Centre for Programmable Materials, School of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore 2Materials Science and Technology Division, Oak Ridge National Lab, Oak Ridge Tennessee 37831, USA 3Department of Physics and Astronomy, Vanderbilt University, Nashville, TN 37235, USA 4National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba 305-8565, Japan 5Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China 6Division of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore 7Department of Physics, National Tsing Hua University, Hsinchu 30013, Taiwan 8Centre for Advanced 2D Materials and Graphene Research Centre, National University of Singapore, Singapore 117546 9Department of Physics, National University of Singapore, Singapore 117542 10Institute of Physics, Academia Sinica, Taipei 11529, Taiwan 11Centre for Disruptive Photonic Technologies, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore 12Collaborative Innovation Center of Quantum Matter, Beijing 100871, China 13Centre for Micro-/Nano-electronics (NOVITAS), School of Electrical & Electronic Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore 14CINTRA CNRS/NTU/THALES, UMI 3288, Research Techno Plaza, 50 Nanyang Drive, Border X Block, Level 6, Singapore 637553, Singapore † These authors contributed equally to this work. -

Surface and Interface Characterization of 2D Materials: Transition Metal Dichalcogenide and Black Phosphorous
SURFACE AND INTERFACE CHARACTERIZATION OF 2D MATERIALS: TRANSITION METAL DICHALCOGENIDE AND BLACK PHOSPHOROUS by Hui Zhu APPROVED BY SUPERVISORY COMMITTEE: ___________________________________________ Dr. Robert M. Wallace, Chair ___________________________________________ Dr. Christopher L. Hinkle ___________________________________________ Dr. Jiyoung Kim ___________________________________________ Dr. Kyeongjae Cho Copyright 2017 Hui Zhu All Rights Reserved Dedicated to my husband and my parents SURFACE AND INTERFACE CHARACTERIZATION OF 2D MATERIALS: TRANSITION METAL DICHALCOGENIDE AND BLACK PHOSPHOROUS by HUI ZHU, BS, MS DISSERTATION Presented to the Faculty of The University of Texas at Dallas in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN MATERIALS SCIENCE AND ENGINEERING THE UNIVERSITY OF TEXAS AT DALLAS December 2017 ACKNOWLEDGMENTS First of all, I’d like to thank my supervisor, Dr. Robert M. Wallace, for his continuous and tremendous support of my master’s and Ph.D. research, for his patience, motivation, and immense knowledge. His diligent working attitude and enthusiasm for science establish a great standard of learning for every researcher in our group, including me. An open, friendly, and positive working environment is created, and it is my pleasure to work and study there. I would also like to acknowledge my committee professors, Dr. Jiyoung Kim, Dr. Kyeongjae Cho, and Dr. Christopher L. Hinkle for their stimulating instruction and the supportive research environment they provided. Grateful appreciation is also delivered to my colleague Mr. Qingxiao Wang and his supervisor Dr. Moon J. Kim for their extensive STEM collaborations included in this work. My acknowledgment to all my colleagues working together on 2D materials at UTD, including postdoctoral researchers: Dr. -

Intrinsic Phonon Bands in High-Quality Monolayer T' Molybdenum Ditelluride
Intrinsic Phonon Bands in High-Quality Monolayer T’ Molybdenum Ditelluride Shao-Yu Chen†, Carl H. Naylor§, Thomas Goldstein†, A.T. Charlie Johnson§, and Jun Yan†* †Department of Physics, University of Massachusetts, Amherst, Massachusetts 01003, United States §Department of Physics and Astronomy, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States *Corresponding Author: Jun Yan. Tel: (413)545-0853 Fax: (413)545-1691 E-mail: [email protected] 1 Abstract The topologically nontrivial and chemically functional distorted octahedral (T’) transition metal dichalcogenides (TMDCs) are a type of layered semimetal that has attracted significant recent attention. However, the properties of monolayer (1L) T’-TMDC, a fundamental unit of the system, is still largely unknown due to rapid sample degradation in air. Here we report that well-protected 1L CVD T'-MoTe2 exhibits sharp and robust intrinsic Raman bands, with intensities about one order of magnitude stronger than those from bulk T'-MoTe2. The high-quality samples enabled us to reveal the set of all nine even- parity zone-center optical phonons, providing reliable fingerprints for the previously elusive crystal. By performing light polarization and crystal orientation resolved scattering analysis, we can effectively distinguish the intrinsic modes from Te-metalloid-like modes A (~122 cm-1) and B (~141 cm-1) which are related to the sample degradation. Our studies offer a powerful non-destructive method for assessing sample quality and for monitoring sample degradation in situ, representing a solid advance in understanding the fundamental properties of 1L-T'-TMDCs. Keywords Transition metal dichalcogenide, monolayer T’ molybdenum ditelluride, Raman scattering, anisotropy, topological insulator, crystal quality 2 Molybdenum and tungsten based transition metal dichalcogenides (TMDCs) are a class of layered materials hosting versatile phases and polymorphs that exhibit fascinating physical, chemical and 1–5 topological properties. -

Draft Report on Carcinogens Monograph on Antimony Trioxide
Draft Report on Carcinogens Monograph on Antimony Trioxide Peer-Review Draft November 29, 2017 Office of the Report on Carcinogens Division of the National Toxicology Program National Institute of Environmental Health Sciences U.S. Department of Health and Human Services This information is distributed solely for the purpose of pre-dissemination peer review under applicable information quality guidelines. It has not been formally distributed by the National Toxicology Program. It does not represent and should not be construed to represent any NTP determination or policy. This Page Intentionally Left Blank Peer-Review Draft RoC Monograph on Antimony Trioxide 11/29/17 Foreword The National Toxicology Program (NTP) is an interagency program within the Public Health Service (PHS) of the Department of Health and Human Services (HHS) and is headquartered at the National Institute of Environmental Health Sciences of the National Institutes of Health (NIEHS/NIH). Three agencies contribute resources to the program: NIEHS/NIH, the National Institute for Occupational Safety and Health of the Centers for Disease Control and Prevention (NIOSH/CDC), and the National Center for Toxicological Research of the Food and Drug Administration (NCTR/FDA). Established in 1978, the NTP is charged with coordinating toxicological testing activities, strengthening the science base in toxicology, developing and validating improved testing methods, and providing information about potentially toxic substances to health regulatory and research agencies, scientific and medical communities, and the public. The Report on Carcinogens (RoC) is prepared in response to Section 301 of the Public Health Service Act as amended. The RoC contains a list of identified substances (i) that either are known to be human carcinogens or are reasonably anticipated to be human carcinogens and (ii) to which a significant number of persons residing in the United States are exposed. -

Barkhausen Effect in the First Order Structural Phase Transition in Type-II Weyl Semimetal Mote2
2D Materials PAPER Barkhausen effect in the first order structural phase transition in type-II Weyl semimetal MoTe2 To cite this article: Chuanwu Cao et al 2018 2D Mater. 5 044003 View the article online for updates and enhancements. This content was downloaded from IP address 162.105.145.73 on 18/10/2018 at 06:09 IOP 2D Materials 2D Mater. 5 (2018) 044003 https://doi.org/10.1088/2053-1583/aae0de 2D Mater. 5 PAPER 2018 Barkhausen effect in the first order structural phase transition RECEIVED © 2018 IOP Publishing Ltd 30 June 2018 in type-II Weyl semimetal MoTe2 REVISED 18 August 2018 2D MATER. Chuanwu Cao1,2 , Xin Liu1,2, Xiao Ren1,2, Xianzhe Zeng1,2, Kenan Zhang2,3, Dong Sun1,2 , Shuyun Zhou2,3 , ACCEPTED FOR PUBLICATION 4 1,2 1,2 12 September 2018 Yang Wu ,i Yuan L and Jian-Hao Chen 1 044003 PUBLISHED International Center for Quantum Materials, School of Physics, Peking University, NO. 5 Yiheyuan Road, Beijing, 100871, 17 October 2018 People’s Republic of China 2 Collaborative Innovation Center of Quantum Matter, Beijing 100871, People’s Republic of China 3 C Cao et al State Key Laboratory of Low Dimensional Quantum Physics, Department of Physics, Tsinghua University, Beijing 100084, People’s Republic of China 4 Tsinghua-Foxconn Nanotechnology Research Center, Tsinghua University, Beijing 100084, People’s Republic of China E-mail: [email protected] Keywords: 2D materials, Barkhausen effect, structural phase transition, MoTe2 Supplementary material for this article is available online 2DM Abstract We report the first observation of the non-magnetic Barkhausen effect in van der Waals layered 10.1088/2053-1583/aae0de crystals, specifically, during transitions between the Td and 1T′ phases in type-II Weyl semimetal MoTe 2. -
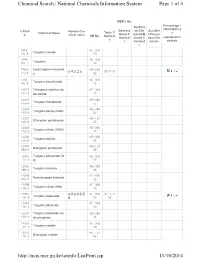
The List of Tungsten and Its Related Compounds
Chemical Search | National Chemicals Information System Page 1 of 4 NIER's No. Percentage i Restrict nformation o Observa ed Che Acciden CAS N Korean Che Toxic C Chemical Name tional C micals/B t Precau f o. mical name KE No. hemical hemical anned C tion Che regulated ch s s hemical micals emicals s 1314- KE-350 35-8 Tungsten trioxide 23 7440- KE-350 33-7 Tungsten 00 7759- Lead tungsten tetraoxid KE-219 납화합물질 97-1-9 01-5 e 52 7783- KE-350 82-6 Tungsten hexafluoride 12 10527 Tritungsten diyttrium do KE-349 -41-0 decaoxide 42 12007 KE-350 -09-9 Tungsten monoboride 15 12007 KE-350 -10-2 Tungsten boride (W2B) 02 12007 KE-127 -98-6 Ditungsten pentaboride 47 12033 KE-350 -72-6 Tungsten nitride (W2N) 17 12036 KE-350 -22-5 Tungsten dioxide 06 12036 KE-127 -84-9 Ditungsten pentaoxide 48 12037 Tungsten phosphide (W KE-350 -70-6 P) 19 12039 KE-350 -88-2 Tungsten disilicide 08 12039 KE-280 -95-1 Pentatungsten trisilicide 22 12058 KE-350 -38-7 Tungsten nitride (WN) 16 12067 셀레늄화합물 KE-350 97-1-1 -46-8 Tungsten diselenide 질 07 34 12067 KE-350 -76-4 Tungsten ditelluride 10 12067 Tungsten hydroxide oxi KE-350 -99-1 de phosphate 14 12070 KE-350 -12-1 Tungsten carbide 03 12070 KE-127 -13-2 Ditungsten carbide 46 http://ncis.nier.go.kr/totinfo/ListPrint.jsp 11/10/2014 Chemical Search | National Chemicals Information System Page 2 of 4 12138 Tungsten disulfide KE-350 -09-9 09 12228 KE-350 -69-2 Tungsten diboride 04 12398 Diammonium tetratungs KE-098 -61-7 ten tridecaoxide 29 12608 Tetratungsten undecao KE-337 -26-3 xide 11 12627 KE-350 -39-3 Tungsten boride -

A Review of the Characteristics, Synthesis, and Thermodynamics of Type-II Weyl Semimetal Wte2
materials Review A Review of the Characteristics, Synthesis, and Thermodynamics of Type-II Weyl Semimetal WTe2 Wenchao Tian, Wenbo Yu * ID , Xiaohan Liu, Yongkun Wang and Jing Shi School of Electro-Mechanical Engineering, Xidian University, Number 2 Taibai South Road, Xi’an 710071, China; [email protected] (W.T.); [email protected] (X.L.); [email protected] (Y.W.); [email protected] (J.S.) * Correspondence: [email protected]; Tel.: +86-29-8820-2954 Received: 4 June 2018; Accepted: 6 July 2018; Published: 10 July 2018 Abstract: WTe2 as a candidate of transition metal dichalcogenides (TMDs) exhibits many excellent properties, such as non-saturable large magnetoresistance (MR). Firstly, the crystal structure and characteristics of WTe2 are introduced, followed by a summary of the synthesis methods. Its thermodynamic properties are highlighted due to the insufficient research. Finally, a comprehensive analysis and discussion are introduced to interpret the advantages, challenges, and future prospects. Some results are shown as follows. (1) The chiral anomaly, pressure-induced conductivity, and non-saturable large MR are all unique properties of WTe2 that attract wide attention, but it is also a promising thermoelectric material that holds anisotropic ultra-low thermal conductivity −1 −1 (0.46 W·m ·K ). WTe2 is expected to have the lowest thermal conductivity, owing to the heavy atom mass and low Debye temperature. (2) The synthesis methods influence the properties significantly. Although large-scale few-layer WTe2 in high quality can be obtained by many methods, the preparation has not yet been industrialized, which limits its applications. (3) The thermodynamic properties of WTe2 are influenced by temperature, scale, and lattice orientations.