Annulation of Non-Conjugated Alkenes Via Directed Nucleopalladation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oxidative Addition & Reductive Elimination
3/1/2021 Oxidative Addition & Reductive Elimination Robert H. Crabtree: Pages 159 – 182 and 235 - 266 1 Oxidative Addition Change in Example Group configuration • Oxidative addition is a key step in many transition-metal catalyzed reactions 10 8 I III • Basic reaction: d d Cu Cu 11 X X Pd0 PdII 10 LnM + LnM Pt0 PtII 10 Y Y d8 d6 PdII PdIV 10 • The new M-X and M-Y bonds are formed using the electron pair of the X-Y PtII PtIV 10 bond and one metal-centered lone pair IrI IrIII 9 RhI RhIII 9 • The metal goes up in oxidation state (+2), X-Y formally gets reduced to X-, Y- d6 d4 ReI ReIII 7 • The ease of addition (or elimination) can be tuned by the electronic and 0 II steric properties of the ancillary ligands Mo Mo 6 • OA favored by strongly e-donating L d4 d2 MoII MoIV 6 • The most common applications involve: a) Late transition metals (platinum metals: Ru, Rh, Pd, Ir, Pt) - not d7 d6 2CoII 2CoIII 9 too sensitive to O2 and H2O; routinely used in organic synthesis b) C-Halogen, H-H or Si-H bonds d4 d3 2CrII 2CrIII 6 • Common for transition metals, rare for main-group metals but: Grignard reagents! 2 1 3/1/2021 Oxidative addition – concerted mechanism Concerted addition - mostly with non-polar X-Y bonds X X X LnM + LnM LnM Y Y Y A Cr(CO)5: • H2, silanes, alkanes, ... coordinatively Cr(PMe3)5: • Arene C-H bonds more reactive than alkane C-H bonds unsaturated, but metal- centered lone pairs not phosphines are better • H-H, C-H strong bond, but M-H and M-C bonds can be very available donors, weaker acceptors full oxidative -
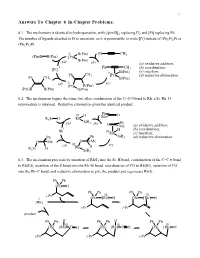
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine
pubs.acs.org/JACS Article Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine Cross-Coupling To Generate Quaternary Carbons Jesuś G. Estrada, Wendy L. Williams, Stephen I. Ting, and Abigail G. Doyle* Cite This: J. Am. Chem. Soc. 2020, 142, 8928−8937 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: We previously reported the development of an electron-deficient olefin (EDO) ligand, Fro-DO, that promotes the generation of quaternary carbon centers via Ni-catalyzed Csp3− Csp3 cross-coupling with aziridines. By contrast, electronically and structurally similar EDO ligands such as dimethyl fumarate and electron-deficient styrenes afford primarily β-hydride elimination side reactivity. Only a few catalyst systems have been identified that promote the formation of quaternary carbons via Ni-catalyzed Csp3−Csp3 cross-coupling. Although Fro-DO represents a promis- ing ligand in this regard, the basis for its superior performance is not well understood. Here we describe a detailed mechanistic study of the aziridine cross-coupling reaction and the role of EDO ligands in facilitating Csp3−Csp3 bond formation. This analysis reveals that cross-coupling proceeds by a Ni0/II cycle with a NiII azametallacyclobutane catalyst resting state. Turnover-limiting C−C reductive elimination occurs from a spectroscopically observable NiII-dialkyl intermediate bound to the EDO. Computational analysis shows that Fro-DO accelerates turnover limiting reductive elimination via LUMO lowering. However, it is no more effective than dimethyl fumarate at reducing the barrier to Csp3− Csp3 reductive elimination. Instead, Fro-DO’s unique reactivity arises from its ability to associate favorably to NiII intermediates. -

Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry
molecules Review Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry Felipe Cesar Sousa e Silva, Anthony F. Tierno and Sarah E. Wengryniuk * Department of Chemistry, Temple University, 1901 N. 13th St., Philadelphia, PA 19122, USA; [email protected] (F.C.S.S.); [email protected] (A.F.T.) * Correspondence: [email protected]; Tel.: +1-215-204-0360 Academic Editor: Margaret A. Brimble Received: 29 March 2017; Accepted: 8 May 2017; Published: 12 May 2017 Abstract: Over the last 20 years, high valent metal complexes have evolved from mere curiosities to being at the forefront of modern catalytic method development. This approach has enabled transformations complimentary to those possible via traditional manifolds, most prominently carbon-heteroatom bond formation. Key to the advancement of this chemistry has been the identification of oxidants that are capable of accessing these high oxidation state complexes. The oxidant has to be both powerful enough to achieve the desired oxidation as well as provide heteroatom ligands for transfer to the metal center; these heteroatoms are often subsequently transferred to the substrate via reductive elimination. Herein we will review the central role that hypervalent iodine reagents have played in this aspect, providing an ideal balance of versatile reactivity, heteroatom ligands, and mild reaction conditions. Furthermore, these reagents are environmentally benign, non-toxic, and relatively inexpensive compared to other inorganic oxidants. We will cover advancements in both catalysis and high valent complex isolation with a key focus on the subtle effects that oxidant choice can have on reaction outcome, as well as limitations of current reagents. Keywords: hypervalent iodine; oxidation; oxidant; redox; high valent; high oxidation state; catalysis 1. -

Reversible C–C Bond Formation Using Palladium Catalysis
Reversible C–C Bond Formation Using Palladium Catalysis Austin Marchese University of Toronto https://orcid.org/0000-0002-3090-2748 Bijan Mirabi University of Toronto https://orcid.org/0000-0002-4852-3439 Colton Johnson University of Toronto https://orcid.org/0000-0002-0905-1248 Mark Lautens ( [email protected] ) University of Toronto https://orcid.org/0000-0002-0179-2914 Article Keywords: C-C bonds, reactions, metal-catalyzed reactions Posted Date: May 13th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-426770/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License 1 2 Reversible C–C Bond Formation Using Palladium Catalysis 3 Authors: Austin D. Marchese1†, Bijan Mirabi1†, Colton E. Johnson1, Mark Lautens1* 4 Affiliations: 5 1Department of Chemistry, Davenport Chemical Laboratories, University of Toronto, Toronto, 6 Ontario M5S 3H6, Canada. 7 *Correspondence to: Department of Chemistry, Davenport Chemical Laboratories, University of 8 Toronto, Toronto, Ontario M5S 3H6, Canada; orcid.org/0000-0002-0179- 2914; Email: 9 [email protected]. 10 †These authors contributed equally. 11 Abstract: A widely appreciated principle is that all reactions are fundamentally reversible. 12 Observing reversible transition metal-catalyzed reactions, particularly those that include the 13 cleavage of C–C bonds, are more challenging. The development of the palladium- and nickel- 14 catalyzed carboiodination reactions afforded access to the syn- and anti-diastereomers of the 15 iodo-dihydroisoquinolone products. Using these substrates, an extensive study investigating the 16 catalytic reversibility of the C–C bond formation using a different palladium catalyst was 17 undertaken. -

• Oxidative Addition • Reductive Elimination • Migratory Insertion • Β - Hydrogen Elimination AJELIAS L7-S2 Oxidative Addition
AJELIAS L7-S1 Unique reactions in organometallic chemistry • Oxidative Addition • Reductive Elimination • Migratory Insertion • β - Hydrogen Elimination AJELIAS L7-S2 Oxidative addition When addition of ligands is accompanied by oxidation of the metal, it is called an oxidative addition reaction LnM+XY Ln(X)(Y)M dn dn-2 OX state of metal increases by 2 units H H2 oxidative Ph3P PPh3 addition H PPh3 Coordination number increases by 2 units Rh Rh Ph3P Cl Ph3P Cl PPh3 2 new anionic ligands are added to the metal Rh+1 Rh+3 Requirements for oxidative addition • availability of nonbonded electron density on the metal, • two vacant coordination sites on the reacting complex (LnM), that is, the complex must be coordinatively unsaturated, • a metal with stable oxidation states separated by two units; the higher oxidation state must be energetically accessible and stable. AJELIAS L7-S3 Examples of Oxidative addition : Cis or trans ? Cl Cl PPh3 Ir 18E Cl2 Ph3P CO Cl O Cl PPh3 O2 O PPh3 Ir Ir Ph3P CO Ph3P CO 16E Cl Me Me MeI Cl PPh 3 I PPh3 Ir Ir Ph P CO 3 Ph3P CO I Cl Homonuclear systems (H2, Cl2, O2, C2H2) Cis Heteronuclear systems (MeI) Cis or trans AJELIAS L7-S4 An important step in many homogeneous catalytic cycles Hydrogenation of alkenes- Wilkinson catalyst H H2 oxidative Ph3P PPh3 addition H PPh3 Rh Rh Often thefirststepofmechanism Ph3P Cl Ph3P Cl PPh3 Rh+1 Rh+3 Methanol to acetic acid conversion- Cativa process CH3 I CO CH3I I CO Ir Ir I CO I CO Ir+1 Ir+3 I Pd catalyzed Cross coupling of Ar-B(OH)2 and Ar-X – Suzuki Coupling Br Br Ph3P Pd -

Cross-Coupling Reactions: A
Joesphine Eshon & Prithvi Vangal 1 11th November 2014 . Introduction . Cross-coupling reactions: A. Negishi* Reaction B. Heck* Reaction C. Stille Reaction D. Suzuki* Reaction E. Sonogashira Reaction F. Buchwald-Hartwig Reaction . Summary 2 . Reactions that form (usually) carbon-carbon bonds between complex fragments . Typically use a transition metal catalyst and an organometallic precursor . Most involve a “transmetallation step” . Transmetallation: Transfer of alkyl group from one metal to another . Typical trend: Can transfer from more electropositive to less electropositive metals 3 Pd Catalyst . General reaction scheme: R-X + R . R: Lacks a β hydrogen attached to an sp3 carbon. (Aryl/Benzyl/Vinyl/Allyl) . X: Typically Cl, Br, I, Otf . Regioselectivity and rates are determined by steric hindrance at the alkene CH CHR RCH~ RRC 2 > RRC > HCR > H2C H2C H2C HCR 4 Spessard and Meissler, Organometallic Chemistry . Palladium is in the 0 oxidation state in the active catalyst : PPh3 Pd Ph3P PPh3 e PdCl2 0 . The palladium can be reduced in situ : Pd PPh3 Pd(OAc)2 . Preferred solvent is DMF . Increases rate, lowers temp. to from 800 C : - n Bu N+ ( )4 + KHCO3 . Two catalytic cycles are possible depending on the reaction conditions 5 Spessard and Meissler, Organometallic Chemistry Oxidative Addition: Pd(0) Pd(+2) d10 d8 14 e 16 e 6 Insertion: Pd(+2) Pd(+2) d8 d8 16 e 16 e 7 Hydride elimination: Pd(+2) Pd(+2) d8 d8 16 e 16 e 8 Reductive elimination: Pd(+2) Pd(0) d8 d10 16 e 14 e 9 Example of an intramolecular Heck reaction 10 BINAP . By preventing β elimination from the new sp3 center, an asymmetric product may be formed . -

Reductive Elimination of Alkylamines and Ethers: Reactions of Bisphosphine-Ligated Palladium(Ii) Complexes
REDUCTIVE ELIMINATION OF ALKYLAMINES AND ETHERS: REACTIONS OF BISPHOSPHINE-LIGATED PALLADIUM(II) COMPLEXES BY SETH L. MARQUARD DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate College of the University of Illinois at Urbana-Champaign 2011 Urbana, Illinois Doctoral committee: Professor John F. Hartwig, Chair Professor Thomas B. Rauchfuss Professor Patricia A. Shapley Professor M. Christina White Abstract The reductive elimination reactions detailed in this dissertation provide experimental insight into the mechanism of reductive elimination to form the C(sp3)-N bond of benzylamines and the C(sp3)-O bond of benzyl ethers. The stereochemical outcome of the reaction indicates an ionic pathway, but the process lacks many of the effects of electronic and solvent perturbations that typically signal an ionic intermediate. We propose that reductive elimination from benzylpalladium(II) amido and aryloxide complexes occurs by dissociation of the amido or aryloxide ligand, followed by nucleophilic attack on the benzyl ligand. The proposed ionic mechanism is more akin to the reductive elimination reactions that occur from high-valent Pt(IV) and Ni(III) complexes than reductive elimination reactions that occur from other Pd(II) complexes. Our data indicate that substantial differences exist between reductive eliminations to form the C(sp3) bonds in ethers and amines from palladium(II). We prepared alkylpalladium(II) amido complexes to study the C(sp3)-N reductive elimination reaction from complexes containing a non-benzylic hydrocarbyl ligand. We investigated a series of alkylpalladium amido complexes and observed reductive elimination occurs from bisphosphine-ligated neopentylpalladium amido complexes in low yield. -

1 Stille Polycondensation: a Versatile Synthetic Approach to Functional Polymers
1 1 Stille Polycondensation: A Versatile Synthetic Approach to Functional Polymers Tianyue Zheng, Alexander M. Schneider, and Luping Yu 1.1 Introduction The development of functional polymers is a very active research field that covers every aspects of our lives and has had huge impact on human society due to their applications in many cutting-edge technologies, such as energy conversion and storage, electronic devices, biotechnology, and health care, to name a few [1]. Scientists from different disciplines have invented numerous new materials for those purposes. Integral to these efforts is the development of efficient, versatile, and scalable synthesis techniques, which in turn enable the development of new functional materials. Thus, new synthetic methodologies are always a critical research topic that is actively pursued. A large number of recent advances can be cited to support this view, such as ring-opening metathesis polymerization (ROMP), atom transfer radical polymerization (ATRP, a type of “living” radical polymerization), and controlled Ziegler–Natta polymerization [2, 3]. Most recently, polycondensations based on transition metal-catalyzed CC bond formation reactions have emerged as important methodologies for synthesis of electro-optic materials containing large systems. These reactions include Stille, Suzuki, Negishi, Heck, and so on [4–7]. The Stille reaction is one of the best methods for the synthesis of organic functional materials due to its excellent compatibility with various functional groups and high reaction yield. The most attractive application of the Stille coupling reaction is in the synthesis of conjugated, polyaromatic semiconducting materials, which are an important class of materials for organic electronics. These materials exhibit good solubility in various sol- vents, which allows them to be fabricated into devices using inexpensive solution-phase printing techniques [8]. -

Rationalization of the Mechanism of in Situ Pd(0) Formation for CrossCoupling Reactions from Novel Unsymmetrical Pincer Palladacycles Using DFT Calculations
Rationalization of the mechanism of in situ Pd(0) formation for cross-coupling reactions from novel unsymmetrical pincer palladacycles using DFT calculations Article (Accepted Version) Boonseng, Sarote, Roffe, Gavin W, Targema, Msugh, Spencer, John and Cox, Hazel (2017) Rationalization of the mechanism of in situ Pd(0) formation for cross-coupling reactions from novel unsymmetrical pincer palladacycles using DFT calculations. Journal of Organometallic Chemistry, 845. pp. 71-81. ISSN 0022-328X This version is available from Sussex Research Online: http://sro.sussex.ac.uk/id/eprint/67061/ This document is made available in accordance with publisher policies and may differ from the published version or from the version of record. If you wish to cite this item you are advised to consult the publisher’s version. Please see the URL above for details on accessing the published version. Copyright and reuse: Sussex Research Online is a digital repository of the research output of the University. Copyright and all moral rights to the version of the paper presented here belong to the individual author(s) and/or other copyright owners. To the extent reasonable and practicable, the material made available in SRO has been checked for eligibility before being made available. Copies of full text items generally can be reproduced, displayed or performed and given to third parties in any format or medium for personal research or study, educational, or not-for-profit purposes without prior permission or charge, provided that the authors, title and full bibliographic details are credited, a hyperlink and/or URL is given for the original metadata page and the content is not changed in any way. -

The Stille Reaction Chem 115
Myers The Stille Reaction Chem 115 Recent Reviews: • Oxidative addition initally gives a cis complex that can rapidly isomerize to the trans isomer: Williams, R. Org. Synth. 2011, 88, 197–201. Selig, R.; Schollmeyer, D.; Albrecht, W.; Laufer, S. Tetrahedron 2011, 67, 9204–9213. R L PdL2 fast Tietze, L. F.; Dufert, A. Pure Appl. Chem., 2010, 82, 1375–1392. R–I L Pd I R Pd I L L Generalized Cross-Coupling: cis trans R–X R'–M catalyst R–R' M–X Casado, A. L.; Espinet, P. Organometallics 1998, 17, 954–959. • !-hydride elimination can be a serious side reaction within alkyl palladium intermediates. This typically requires a syn coplanar alignment of hydride and palladium: Typically: • R and R' are sp2–hybridized • M = Sn, B, Zr, Zn • X = I, OSO2CF3, Br, Cl • catalyst = Pd (sometimes Ni) H Pd(II)L2X + HPd(II)L2X Mechanism: • A specific example: • Oxidative-addition and reductive-elimination steps occur with retention of configuration for 2 Pd catalyst sp -hybridized substrates. p-Tol–Br + n-Bu3Sn–Ph p-Tol–Ph + n-Bu3Sn–Br Pd(II) • Transmetalation is proposed to be the rate-determining step with most substrates. • Relative order of ligand transfer from Sn: p-Tol–Ph Pd(0)Ln p-Tol–Br alkynyl > alkenyl > aryl > allyl = benzyl > "-alkoxyalkyl > alkyl reductive elimination oxidative addition • Electron-rich and sterically hindered aryl halides undergo slower oxidative addition and are often poor substrates as a result. p-Tol–Pd(II)Lm–Ph p-Tol–Pd(II)Lm–Br • Electron-poor stannanes undergo slower transmetallation and are often poor substrates as n-Bu Sn–Br n-Bu Sn–Ph 3 3 a result. -

A Summary of Organometallic Chemistry Counting Valence Electrons (V.E.) with the Ionic Model 1
A Summary of Organometallic Chemistry Counting valence electrons (v.e.) with the ionic model 1. Look at the total charge of the complex + 2– Ph3P Cl OC CO Rh Fe Co Ph3P PPh3 OC CO charge:0 charge: -2 charge: +1 2. Look at the charge of the ligands (see table in next page) and calculate the formal oxidation state of the metal and therefore the d electrons at the metal center + 2– Ph3P Cl OC CO Rh Fe Co Ph3P PPh3 OC CO Rh+ Fe2- Co3+ d8 d10 d6 3. Add the electrons coming from all ligands + 2– Ph3P Cl OC CO Rh Fe Co Ph3P PPh3 OC CO 1 x 2e- (Cl) 4 x 2e- (CO) 2 x 6e- (Cp) 3 x 2e- (PPh3) TOT 8 e- TOT 8 e- TOT 12 e- 4. Add the electrons of both metal and ligands to get the valence electrons (v.e.) + 2– Ph3P Cl OC CO Rh Fe Co Ph3P PPh3 OC CO 16 v.e. 18 v.e. 18 v.e. The 18-electron rule The rule helps us to decide whether a complex is likely to be stable. To have a stable transition metal we need to fill all s, p, and d orbitals. These are a total of 9 orbitals, which means 18 electrons. The rule is similar to the 8-electron rule of main group elements. Exceptions to the 18-electrons rule - Early transition metal complexes and/or complexes with bulky ligands are stable with less than 18 v.e. mainly for steric reasons. - Complexes with many π-donor ligands (see below) are stable even with less than 18 v.e.