Reductive Elimination of Alkylamines and Ethers: Reactions of Bisphosphine-Ligated Palladium(Ii) Complexes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oxidative Addition & Reductive Elimination
3/1/2021 Oxidative Addition & Reductive Elimination Robert H. Crabtree: Pages 159 – 182 and 235 - 266 1 Oxidative Addition Change in Example Group configuration • Oxidative addition is a key step in many transition-metal catalyzed reactions 10 8 I III • Basic reaction: d d Cu Cu 11 X X Pd0 PdII 10 LnM + LnM Pt0 PtII 10 Y Y d8 d6 PdII PdIV 10 • The new M-X and M-Y bonds are formed using the electron pair of the X-Y PtII PtIV 10 bond and one metal-centered lone pair IrI IrIII 9 RhI RhIII 9 • The metal goes up in oxidation state (+2), X-Y formally gets reduced to X-, Y- d6 d4 ReI ReIII 7 • The ease of addition (or elimination) can be tuned by the electronic and 0 II steric properties of the ancillary ligands Mo Mo 6 • OA favored by strongly e-donating L d4 d2 MoII MoIV 6 • The most common applications involve: a) Late transition metals (platinum metals: Ru, Rh, Pd, Ir, Pt) - not d7 d6 2CoII 2CoIII 9 too sensitive to O2 and H2O; routinely used in organic synthesis b) C-Halogen, H-H or Si-H bonds d4 d3 2CrII 2CrIII 6 • Common for transition metals, rare for main-group metals but: Grignard reagents! 2 1 3/1/2021 Oxidative addition – concerted mechanism Concerted addition - mostly with non-polar X-Y bonds X X X LnM + LnM LnM Y Y Y A Cr(CO)5: • H2, silanes, alkanes, ... coordinatively Cr(PMe3)5: • Arene C-H bonds more reactive than alkane C-H bonds unsaturated, but metal- centered lone pairs not phosphines are better • H-H, C-H strong bond, but M-H and M-C bonds can be very available donors, weaker acceptors full oxidative -

Studies of a Dirhodium Tetraphosphine Catalyst for Hydroformylation And
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2012 Studies of a dirhodium tetraphosphine catalyst for hydroformylation and aldehyde-water shift ac talysis Aaron Rider Barnum Louisiana State University and Agricultural and Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Chemistry Commons Recommended Citation Barnum, Aaron Rider, "Studies of a dirhodium tetraphosphine catalyst for hydroformylation and aldehyde-water shift catalysis" (2012). LSU Doctoral Dissertations. 3998. https://digitalcommons.lsu.edu/gradschool_dissertations/3998 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. STUDIES OF A DIRHODIUM TETRAPHOSPHINE CATALYST FOR HYDROFORMYLATION AND ALDEHYDE-WATER SHIFT CATALYSIS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College In partial fulfillment of the Requirements for the degree of Doctor of Philosophy In The Department of Chemistry by Aaron Rider Barnum B.S. Loyola University New Orleans, 2007 December 2012 ACKNOWLEDGEMENTS I would like to thank my family, for without their encouragements and support I would not be where I am today. To my parents, Otis and Cindy Barnum, thank you for everything throughout the years. To my grandmother Teruko, you are responsible for two things I hold very dear to my heart: inspiring me to become the scientist and chemist I am today and also for keeping me in touch with my Japanese heritage. -

Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis
UvA-DARE (Digital Academic Repository) Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis Lankelma, M.; Vreeken, V.; Siegler, M.A.; van der Vlugt, J.I. DOI 10.3390/inorganics7030028 Publication date 2019 Document Version Final published version Published in Inorganics License CC BY Link to publication Citation for published version (APA): Lankelma, M., Vreeken, V., Siegler, M. A., & van der Vlugt, J. I. (2019). Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis. Inorganics, 7(3), [28]. https://doi.org/10.3390/inorganics7030028 General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:02 Oct 2021 inorganics Article Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis Marianne Lankelma 1, Vincent Vreeken 1, Maxime A. -

Transition Metal Complexes of a Novel Tetradentate Phosphine and of a New Diphosphine Ether Mark R
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1991 Transition metal complexes of a novel tetradentate phosphine and of a new diphosphine ether Mark R. Mason Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Inorganic Chemistry Commons Recommended Citation Mason, Mark R., "Transition metal complexes of a novel tetradentate phosphine and of a new diphosphine ether " (1991). Retrospective Theses and Dissertations. 9554. https://lib.dr.iastate.edu/rtd/9554 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfîlm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from ary type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely afreet reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -
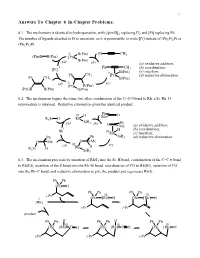
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

Communication
COMMUNICATION DOI: 10.1002/adsc.201500313 Rhodium(I)-Catalyzed Intermolecular Hydroacylation of α- Keto Amides and Isatins with Non-Chelating Aldehydes Kevin G. M. Kou,a,b Lauren E. Longobardi,b and Vy M. Donga* a University of California, Irvine, Department of Chemistry, Natural Sciences I, Irvine, California 92697, United States Fax: (949) 824-8571 Email: [email protected] b University of Toronto, Department of Chemistry, 80 St. George St., Toronto, ON, Canada M5S 3H6 Received: April 9, 2015 Dedicated to Prof. Stephen L. Buchwald on the occasion of his 60th birthday. Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######. Abstract. The application of the bidentate, electron-rich we sought to develop an analogous catalyst to enable bisphosphine ligand, 1,3-bis(dicyclohexyl)phosphine- a more general intermolecular transformation. To propane (dcpp), in rhodium(I)-catalyzed intermolecular study this challenging reaction, α-keto amide 2a was ketone hydroacylation is herein described. Isatins and α- chosen as the model ketone substrate, given its ability keto amides are shown to undergo hydroacylation with a to chelate metal centers.[15,16] In the presence of a variety of non-chelating linear and branched aliphatic bisphosphine ligand, chelation of the 1,2-keto amide aldehydes. Also reported is the synthesis of new bidentate unit with concomitant oxidative addition of a simple chiral phosphine ligands, and their application in aldehyde would prevent decarbonylation while hydroacylation is discussed. directing insertion of the ketone into the Rh(III)- hydride (Figure 1). Keywords: ketone; hydroacylation; rhodium; asymmetric catalysis; P ligands While progress has been made in selective C−H oxidation via transition-metal catalyzed ketone hydroacylation,[1-4] the field is still young compared to that of the related ketone hydrosilylation,[5] olefin [6] [7] hydrogenation, hydroformylation, and hydroacylation[8] transformations. -

Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine
pubs.acs.org/JACS Article Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine Cross-Coupling To Generate Quaternary Carbons Jesuś G. Estrada, Wendy L. Williams, Stephen I. Ting, and Abigail G. Doyle* Cite This: J. Am. Chem. Soc. 2020, 142, 8928−8937 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: We previously reported the development of an electron-deficient olefin (EDO) ligand, Fro-DO, that promotes the generation of quaternary carbon centers via Ni-catalyzed Csp3− Csp3 cross-coupling with aziridines. By contrast, electronically and structurally similar EDO ligands such as dimethyl fumarate and electron-deficient styrenes afford primarily β-hydride elimination side reactivity. Only a few catalyst systems have been identified that promote the formation of quaternary carbons via Ni-catalyzed Csp3−Csp3 cross-coupling. Although Fro-DO represents a promis- ing ligand in this regard, the basis for its superior performance is not well understood. Here we describe a detailed mechanistic study of the aziridine cross-coupling reaction and the role of EDO ligands in facilitating Csp3−Csp3 bond formation. This analysis reveals that cross-coupling proceeds by a Ni0/II cycle with a NiII azametallacyclobutane catalyst resting state. Turnover-limiting C−C reductive elimination occurs from a spectroscopically observable NiII-dialkyl intermediate bound to the EDO. Computational analysis shows that Fro-DO accelerates turnover limiting reductive elimination via LUMO lowering. However, it is no more effective than dimethyl fumarate at reducing the barrier to Csp3− Csp3 reductive elimination. Instead, Fro-DO’s unique reactivity arises from its ability to associate favorably to NiII intermediates. -

Synthesis, Properties, and Coordination Chemistry of T-Bu-Xantphos Ligands
Synthesis, Properties, and Coordination Chemistry of t-Bu-Xantphos Ligands by Melanie Ruth Maria Nelson A thesis submitted to the Victoria University of Wellington in fulfilment of the requirements for the degree of Doctor of Philosophy in Chemistry. Victoria University of Wellington 2015 Abstract This thesis provides an account of research into a group of diphosphine ligands with a rigid xanthene backbone and tert-butyl substituents on the phosphorus atoms. The three ligands have different groups in the bridgehead position of the backbone (CMe2, SiMe2, or S) which change the natural (calculated) bite-angle of the ligand. The coordination chemistry of these t-Bu-xantphos ligands with late-transition metals has been investigated with a focus on metal complexes that may form in catalytic reactions. The three t-Bu-xantphos ligands were synthesised by lithiation of the backbone t using sec-butyllithium/TMEDA and treatment with P Bu2Cl. The natural bite- angles of the Ph-xantphos (111.89–114.18◦) and t-Bu-xantphos (126.80–127.56◦) ligands were calculated using DFT. The bite-angle of the t-Bu-xantphos ligands is larger due to the increased steric bulk of the tert-butyl substituents. The elec- tronic properties of the t-Bu-xantphos ligands were also investigated by synthesis 1 of their phosphine selenides. The values of J PSe (689.1–698.5 Hz) indicate that the t-Bu-xantphos ligands have a higher basicity than Ph-xantphos between PPh2Me and PMe3. The silver complexes, [Ag(t-Bu-xantphos)Cl] and [Ag(t-Bu-xantphos)]BF4 were synthesised with the t-Bu-xantphos ligands. -

Catalysts Play a Major Role in Development by A
Catalysts Play a Major Role in Development By A. E Chiffey Chemicals Dewlnprnent, Precious Metals Dix ision, .joli~isniiWatt he?, Rovston The second Anglo-Dutch Symposium on the key reaction stages of oxidative insertion, Catalysis and Organometallic Chemistry was hydrogen activation and the breaking of two held on the 26th September 1997, in carbon-sulfur bonds. The reactions have so Amsterdam, The Netherlands. The symposium far been performed under hydrogen pressures was attended by more than 80 participants fi-om of 20 atmospheres and at a temperature of 100°C industry and universities in Britain and The for 24 hours. While results from these systems Netherlands. A series of lectures was given on have been encouraging and should help in catalysis and its applications, and there were the development of the next generation of more than thirty poster presentations. This hydrodesulfurisation catalysts, scaling these reac- report highlights some aspects involving the tions to tonnage quantities is as yet impractical. platinum group metals. Catalysts with Bite Towards Cleaner Fuels In chelate complexes with bidentate ligands Sulfur compounds, which occur naturally in the angle between the two donor atoms of the crude oil, give the pollutant sulfur dioxide when bidentate ligand and the metal centre is termed burnt. Legislation, particularly in the U.S.A., is the ‘bite angle’. The importance of the bite angle forcing the maximum sulfur limits in fuels to in a series of complexes, was reported by become progressively lower. This requirement, Professor P. W. N. M. van Leeuwen of the to produce more environmentally acceptable University of Amsterdam, see I1 and 111. -

Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry
molecules Review Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry Felipe Cesar Sousa e Silva, Anthony F. Tierno and Sarah E. Wengryniuk * Department of Chemistry, Temple University, 1901 N. 13th St., Philadelphia, PA 19122, USA; [email protected] (F.C.S.S.); [email protected] (A.F.T.) * Correspondence: [email protected]; Tel.: +1-215-204-0360 Academic Editor: Margaret A. Brimble Received: 29 March 2017; Accepted: 8 May 2017; Published: 12 May 2017 Abstract: Over the last 20 years, high valent metal complexes have evolved from mere curiosities to being at the forefront of modern catalytic method development. This approach has enabled transformations complimentary to those possible via traditional manifolds, most prominently carbon-heteroatom bond formation. Key to the advancement of this chemistry has been the identification of oxidants that are capable of accessing these high oxidation state complexes. The oxidant has to be both powerful enough to achieve the desired oxidation as well as provide heteroatom ligands for transfer to the metal center; these heteroatoms are often subsequently transferred to the substrate via reductive elimination. Herein we will review the central role that hypervalent iodine reagents have played in this aspect, providing an ideal balance of versatile reactivity, heteroatom ligands, and mild reaction conditions. Furthermore, these reagents are environmentally benign, non-toxic, and relatively inexpensive compared to other inorganic oxidants. We will cover advancements in both catalysis and high valent complex isolation with a key focus on the subtle effects that oxidant choice can have on reaction outcome, as well as limitations of current reagents. Keywords: hypervalent iodine; oxidation; oxidant; redox; high valent; high oxidation state; catalysis 1. -

Reversible C–C Bond Formation Using Palladium Catalysis
Reversible C–C Bond Formation Using Palladium Catalysis Austin Marchese University of Toronto https://orcid.org/0000-0002-3090-2748 Bijan Mirabi University of Toronto https://orcid.org/0000-0002-4852-3439 Colton Johnson University of Toronto https://orcid.org/0000-0002-0905-1248 Mark Lautens ( [email protected] ) University of Toronto https://orcid.org/0000-0002-0179-2914 Article Keywords: C-C bonds, reactions, metal-catalyzed reactions Posted Date: May 13th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-426770/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License 1 2 Reversible C–C Bond Formation Using Palladium Catalysis 3 Authors: Austin D. Marchese1†, Bijan Mirabi1†, Colton E. Johnson1, Mark Lautens1* 4 Affiliations: 5 1Department of Chemistry, Davenport Chemical Laboratories, University of Toronto, Toronto, 6 Ontario M5S 3H6, Canada. 7 *Correspondence to: Department of Chemistry, Davenport Chemical Laboratories, University of 8 Toronto, Toronto, Ontario M5S 3H6, Canada; orcid.org/0000-0002-0179- 2914; Email: 9 [email protected]. 10 †These authors contributed equally. 11 Abstract: A widely appreciated principle is that all reactions are fundamentally reversible. 12 Observing reversible transition metal-catalyzed reactions, particularly those that include the 13 cleavage of C–C bonds, are more challenging. The development of the palladium- and nickel- 14 catalyzed carboiodination reactions afforded access to the syn- and anti-diastereomers of the 15 iodo-dihydroisoquinolone products. Using these substrates, an extensive study investigating the 16 catalytic reversibility of the C–C bond formation using a different palladium catalyst was 17 undertaken. -

• Oxidative Addition • Reductive Elimination • Migratory Insertion • Β - Hydrogen Elimination AJELIAS L7-S2 Oxidative Addition
AJELIAS L7-S1 Unique reactions in organometallic chemistry • Oxidative Addition • Reductive Elimination • Migratory Insertion • β - Hydrogen Elimination AJELIAS L7-S2 Oxidative addition When addition of ligands is accompanied by oxidation of the metal, it is called an oxidative addition reaction LnM+XY Ln(X)(Y)M dn dn-2 OX state of metal increases by 2 units H H2 oxidative Ph3P PPh3 addition H PPh3 Coordination number increases by 2 units Rh Rh Ph3P Cl Ph3P Cl PPh3 2 new anionic ligands are added to the metal Rh+1 Rh+3 Requirements for oxidative addition • availability of nonbonded electron density on the metal, • two vacant coordination sites on the reacting complex (LnM), that is, the complex must be coordinatively unsaturated, • a metal with stable oxidation states separated by two units; the higher oxidation state must be energetically accessible and stable. AJELIAS L7-S3 Examples of Oxidative addition : Cis or trans ? Cl Cl PPh3 Ir 18E Cl2 Ph3P CO Cl O Cl PPh3 O2 O PPh3 Ir Ir Ph3P CO Ph3P CO 16E Cl Me Me MeI Cl PPh 3 I PPh3 Ir Ir Ph P CO 3 Ph3P CO I Cl Homonuclear systems (H2, Cl2, O2, C2H2) Cis Heteronuclear systems (MeI) Cis or trans AJELIAS L7-S4 An important step in many homogeneous catalytic cycles Hydrogenation of alkenes- Wilkinson catalyst H H2 oxidative Ph3P PPh3 addition H PPh3 Rh Rh Often thefirststepofmechanism Ph3P Cl Ph3P Cl PPh3 Rh+1 Rh+3 Methanol to acetic acid conversion- Cativa process CH3 I CO CH3I I CO Ir Ir I CO I CO Ir+1 Ir+3 I Pd catalyzed Cross coupling of Ar-B(OH)2 and Ar-X – Suzuki Coupling Br Br Ph3P Pd