Synthesis, Properties, and Coordination Chemistry of T-Bu-Xantphos Ligands
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Powerful Framework for the Chemical Design of Molecular Nanomagnets Yan Duan†,1,2, Joana T
Data mining, dashboards and statistics: a powerful framework for the chemical design of molecular nanomagnets Yan Duan†,1,2, Joana T. Coutinho†,*,1,3, Lorena E. Rosaleny†,*,1, Salvador Cardona-Serra1, José J. Baldoví1, Alejandro Gaita-Ariño*,1 1 Instituto de Ciencia Molecular (ICMol), Universitat de València, C/ Catedrático José Beltrán 2, 46980 Paterna, Spain 2 Department of Chemistry and the Ilse Katz Institute for Nanoscale Science & Technology, Ben-Gurion University of the Negev, Beer Sheva 84105, Israel 3 Centre for Rapid and Sustainable Product Development, Polytechnic of Leiria, 2430-028 Marinha Grande, Portugal *e-mail: [email protected], [email protected], [email protected] Abstract Three decades of intensive research in molecular nanomagnets have brought the magnetic memory in molecules from liquid helium to liquid nitrogen temperature. The enhancement of this operational temperature relies on a wise choice of the magnetic ion and the coordination environment. However, serendipity, oversimplified theories and chemical intuition have played the main role. In order to establish a powerful framework for statistically driven chemical design, we collected chemical and physical data for lanthanide-based nanomagnets to create a catalogue of over 1400 published experiments, developed an interactive dashboard (SIMDAVIS) to visualise the dataset, and applied inferential statistical analysis to it. We found that the effective energy barrier derived from the Arrhenius equation displays an excellent correlation with the magnetic memory, and that among all chemical families studied, only terbium bis-phthalocyaninato sandwiches and dysprosium metallocenes consistently present magnetic memory up to high temperature, but that there are some promising strategies for improvement. -

Studies of a Dirhodium Tetraphosphine Catalyst for Hydroformylation And
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2012 Studies of a dirhodium tetraphosphine catalyst for hydroformylation and aldehyde-water shift ac talysis Aaron Rider Barnum Louisiana State University and Agricultural and Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Chemistry Commons Recommended Citation Barnum, Aaron Rider, "Studies of a dirhodium tetraphosphine catalyst for hydroformylation and aldehyde-water shift catalysis" (2012). LSU Doctoral Dissertations. 3998. https://digitalcommons.lsu.edu/gradschool_dissertations/3998 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. STUDIES OF A DIRHODIUM TETRAPHOSPHINE CATALYST FOR HYDROFORMYLATION AND ALDEHYDE-WATER SHIFT CATALYSIS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College In partial fulfillment of the Requirements for the degree of Doctor of Philosophy In The Department of Chemistry by Aaron Rider Barnum B.S. Loyola University New Orleans, 2007 December 2012 ACKNOWLEDGEMENTS I would like to thank my family, for without their encouragements and support I would not be where I am today. To my parents, Otis and Cindy Barnum, thank you for everything throughout the years. To my grandmother Teruko, you are responsible for two things I hold very dear to my heart: inspiring me to become the scientist and chemist I am today and also for keeping me in touch with my Japanese heritage. -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -

Coordination Chemistry (See Chapter 20, H&S 3Rd Ed.)
Coordination Chemistry (see Chapter 20, H&S 3rd Ed.) Transition metal elements, particularly ions, have a strong tendency to form compounds with Lewis bases. Why? Lewis base: electron pair donor egs. H2O, NH3 n+ Lewis acid: electron pair acceptor egs. M , BH3 ‘low lying’ empty orbitals: lower in energy than HOMO of Lewis case Lewis acidity enhanced by positive charge so TM ions are good Lewis bases 2+ 3+ familiar examples: AlCl3, BF3 from main group and M , M in TM series Type of bonding in these compounds? ‘coordinative’ or ‘dative’ bonds to form Lewis base ‘adducts’ L→M TM-Lewis base ‘adducts’ are often referred to as (i) TM ‘complexes’ OR (ii) coordination complexes NB: still polar covalent bonds – this just defines where the pair of electrons came from Some more definitions: ligand: molecule or ion that can act as a Lewis base to a metal - egs. Cl , PR3, ROR (R = generic term for an alkyl) donor atoms: atoms that are in direct contact with the metal ion O in R2O→M denticity: number of donor atoms per ligand; range from 1 to >6 unidentate bidentate tridentate O H O H2N NH2 2 O O water en diglyme N O O O N N O O N porphin 15-C-5 tetradentate pentadentate chelate: multidentate ligands forms rings; M tend to be very stable, OO especially 5 and 6 member chelate rings acac ‘bite’ or ‘bite angle’: a measure of the span or size of a chelate ligand; depends on M-L distance M OO acac coordination complex MLn generic terms commonly used: L neutral Lewis base (electron pair donor) X anionic donor (eg. -
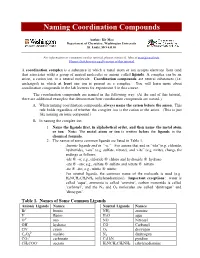
Naming Coordination Compounds
Naming Coordination Compounds Author: Kit Mao Department of Chemistry, Washington University St. Louis, MO 63130 For information or comments on this tutorial, please contact K. Mao at [email protected]. Please click here for a pdf version of this tutorial. A coordination complex is a substance in which a metal atom or ion accepts electrons from (and thus associates with) a group of neutral molecules or anions called ligands. A complex can be an anion, a cation ion, or a neutral molecule. Coordination compounds are neutral substances (i.e. uncharged) in which at least one ion is present as a complex. You will learn more about coordination compounds in the lab lectures for experiment 5 in this course. The coordination compounds are named in the following way. (At the end of this tutorial, there are additional examples that demonstrate how coordination compounds are named.) A. When naming coordination compounds, always name the cation before the anion. This rule holds regardless of whether the complex ion is the cation or the anion. (This is just like naming an ionic compound.) B. In naming the complex ion: 1. Name the ligands first, in alphabetical order, and then name the metal atom or ion. Note: The metal atom or ion is written before the ligands in the chemical formula. 2. The names of some common ligands are listed in Table 1. · Anionic ligands end in “-o.” For anions that end in “-ide”(e.g. chloride, hydroxide), “-ate” (e.g. sulfate, nitrate), and “-ite” (e.g. nirite), change the endings as follows: -ide ® -o; e.g., chloride ® chloro and hydroxide ® hydroxo -ate ® -ato; e.g., sulfate ® sulfato and nitrate ® nitrato -ite ® -ito; e.g., nitrite ® nitrito · For neutral ligands, the common name of the molecule is used (e.g. -

Bridging Ligands
Chapter 9 Coordination Chemistry I: Structure and Isomers 9-1 History 9-2 Nomenclature 9-3 Isomerism 9-4 Coordination Numbers and Structure History What is coordination compound? Coordniantion compounds include compound composed of a metal atom or ion and one or more ligands that formally donate electrons to the metal. More specifically, a transition metal surrounded by neutral molecules or anions with a definite geometry. What is ligand? Ligand can be a atom, ion, and molecules. History Prussian blue (German: Preußischblau or Berliner Blau, in English Berlin blue) is a dark blueWhatpigment is coordinationused in paints and compound? formerly in blueprints. Prussian blue wasCoordination discovered complexesby accident were by pai knownnter Heinrich - although Diesbach not understood in Berlin in 1704-5,in which any sense is why - it since is also the known beginning as Berlin of chemistry, blue. (Diesbach e.g. Prussian was attempting to createblue a paint, Aureolin with ,aand red copperhue.) It vitriolhas several. different chemical names, these being iron(III) ferrocyanide, ferric ferrocyanide, iron(III) hexacyanoferrate(II), and ferricThe hexacyanoferrate. key breakthrough Commonly occurred when and c Alfredonveniently Werner it isproposed, simply called "PB. inter alia, that Co(III) bears six ligands in an octahedral geometry. Aureolin (sometimes called Cobalt Yellow) is a pigment used in oil and watercolor painting. Its color index name is PY40 (40th entry on list of yellow pigments). It was first made in 1851 and its chemical composition is potassium cobaltinitrite. Copper(II) sulfate ("sulphate" in most Commonwealth nations) is the chemical compound with the formula CuSO4. This salt exists as a series of compounds that differ in their degree of hydration. -

Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis
UvA-DARE (Digital Academic Repository) Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis Lankelma, M.; Vreeken, V.; Siegler, M.A.; van der Vlugt, J.I. DOI 10.3390/inorganics7030028 Publication date 2019 Document Version Final published version Published in Inorganics License CC BY Link to publication Citation for published version (APA): Lankelma, M., Vreeken, V., Siegler, M. A., & van der Vlugt, J. I. (2019). Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis. Inorganics, 7(3), [28]. https://doi.org/10.3390/inorganics7030028 General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:02 Oct 2021 inorganics Article Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis Marianne Lankelma 1, Vincent Vreeken 1, Maxime A. -

Complexation
FACULTY OF PHARMACEUTICAL SCIENCES, RAMAUNIVERSITY, KANPUR B.PHARM 3rd SEM PHYSICAL PHARMACEUTICS-I BP302T MR. PEEYUSH Assistant professor Rama university, kanpur Complexation Overview Classification Introduction Metal ion complexes Organic Complexes Inclusion Complexes Methods of Analysis Method of Continuous Variation PH Titration Distribution Method Solubility Method Spectroscopy Learning Objectives 1. Define the three classes of complexes with pharmaceutically relevant examples. 2. Describe chelates, their physically properties, and what differentiates them from organic molecular complexes. 3. Describe the types of forces that hold together organic molecular complexes with examples. 4. Describe the forces in polymer–drug complexes used for drug delivery. 5. Discuss the pharmaceutical applications of cyclodextrins. 6. Describe the methods of analysis of complexes and determine their stoichiometric ratios and stability constants. Classification Introduction Metal ion complexes Organic Complexes Inclusion Complexes INTRODUCTION Complexes are compounds that result from donor–acceptor mechanisms between two or more chemical species. Complexes can be divided broadly into three classes depending the type of the acceptor substance: 1. Metal ion complexes 2. Organic molecular complexes 3. Inclusion complexes Intermolecular forces involved in the formation of complexes: 1. Van der Waals forces. 2. Hydrogen bonds (important in molecular complexes). 3. Coordinate covalence (important in metal complexes). 4. Charge transfer. 5. Hydrophobic interaction. Introduction Types of Complexes Metal Ion Complexes A. Inorganic type B. Chelates C. Olefin type D. Aromatic type II. Organic Molecular Complexes A. Quinhydrone type B. Picric acid type C. Caffeine and other drug complexes D. Polymer type III. Inclusion Compounds A. Channel lattice type B. Layer type C. Clathrates D. Monomolecular type E. -

Asymmetric Desymmetrization of Oxetanes for the Synthesis of Chiral
Angewandte Research Articles Chemie International Edition:DOI:10.1002/anie.201910917 Asymmetric Catalysis German Edition:DOI:10.1002/ange.201910917 Asymmetric Desymmetrization of Oxetanes for the Synthesis of Chiral Tetrahydrothiophenes and Tetrahydroselenophenes Renwei Zhang+,Wengang Guo+,Meng Duan+,K.N.Houk,* and Jianwei Sun* Abstract: Chiral tetrahydrothiophenes and tetrahydroseleno- phenes are highly useful structural units.Described here is anew catalytic asymmetric approach for their synthesis.With asuitable chiral Brønsted acid catalyst, an oxetane desymmet- rization by awell-positioned internal sulfur or selenium nucleophile proceeded efficiently to generate all-carbon qua- ternary stereocenters with excellent enantioselectivities.Taming the sulfur and selenium nucleophile in the form of athioester and selenoester,respectively,iscrucial to the success of this work. This approach also allows the facile synthesis of chiral tetrahydrothiopyrans.Mechanistic studies,including DFT calculations,suggested an intramolecular acyl-transfer path- way.Utilities of the chiral products are also demonstrated. Introduction Figure 1. Useful compounds containing chiral tetrahydrothiophene and tetrahydroselenopheneunits. Chiral organosulfur and organoselenium compounds play important roles in biological processes,organic synthesis,and asymmetric hydrogenation of substituted thiophenes (Sche- medicinal chemistry.[1] In particular, chiral tetrahydrothio- me 1a).[6] However,unfortunately,this elegant process suffers phenes and tetrahydroselenophenes are well-known subunits from alimited scope:good enantioselectivity was only in awide range of natural products and bioactive molecules observed for asmall number of thiophenes bearing specific that exhibit abroad spectrum of important biological aryl substituent at the 2-position. activities (e.g., I–V,Figure 1).[2] Moreover,such chiral units Moreover,the nature of this approach excludes the embedded in arelatively rigid five-membered ring structure possibility of generating aquaternary stereogenic center. -

Sodium-Mediated Magnesiation of Thiophene and Tetrahydrothiophene: Structural Contrasts with Furan and Tetrahydrofuran
Sodium-mediated Magnesiation of Thiophene and Tetrahydrothiophene: Structural Contrasts with Furan and Tetrahydrofuran Victoria L. Blair, Alan R. Kennedy, Robert E. Mulvey and Charles T. O’Hara* V. L. Blair, Dr. A. R. Kennedy, Prof. R. E. Mulvey, Dr. C. T. O’Hara WestCHEM, Department of Pure and Applied Chemistry University of Strathclyde Glasgow, G1 1XL (U.K.) Fax: (+44) 141 548 4822 E-mail: [email protected]; [email protected] Sulfur-containing heterocycles are currently attracting a great deal of interest in several diverse fields. For instance, substituted tetrahydrothiophenes,[1] have received considerable attention due to their extremely wide-ranging chemical and biological applications.[2] These include their use as potent α-glucosidase inhibitors,[3] as an inhibitor of copper amine [4] [5] oxidases and as selective A3 agonists and antagonists. In addition, they have been utilized in chemical transformations, such as catalytic asymmetric epoxidation, catalytic intramolecular cyclopropanation, and asymmetric metal catalysis hydrogenation.[6] From a nanochemical perspective, the adsorption chemistries and physical properties of various thiophenes and tetrahydrothiophenes on gold surfaces have recently come to the fore.[7] Polythiophenes are also key compounds in modern materials research, currently utilised in for example the fabrication of semi-conducting, fluorescent, and electronic and optoelectronic materials.[8]In this work, metallation (exchange of a hydrogen atom with a metal atom) of the parent heterocycles, tetrahydrothiophene (THT) and thiophene is considered. Metallation is one of the most fundamental reactions in modern day synthesis and is a key tool in the preparation of functionalised aromatic and heterocyclic compounds. -

Acceptor Ligands in Stabilizing and Assembling Pt Or Ir Carbonyl Clusters Simona El Afefey
UNIVERSITA’ DEGLI STUDI DI MILANO Scuola di Dottorato in Scienze Chimiche Dipartimento di Chimica PhD in Industrial Chemistry XXVCycle Electron-donor and -acceptor ligands in stabilizing and assembling Pt or Ir carbonyl clusters (Chim 03) Simona El Afefey Supervisor: Prof. ALESSANDRO CERIOTTI Coordinator: Prof. DOMINIQUE ROBERTO A.A. 2011/2012 Simona El Afefey Electron-donor and –acceptor ligands in stabilizing and assembling Pt or Ir carbonyl clusters ________________________________________________________________________________________________ Abstract ............................................................................................................................................... 7 Introduction ..................................................................................................................................... 21 1.1 Carbon Monoxide as ligand .................................................................................................... 21 1.2 General considerations on metal carbonyl clusters ............................................................. 25 1.2.1 Synthetic aspects of metal carbonyl clusters .............................................................. 26 1.2.2 Synthetic methods .......................................................................................................... 28 1.2.3 Direct reductive-carbonylation ................................................................................... 29 1.2.4 Thermal methods .......................................................................................................... -

Transition Metal Complexes of a Novel Tetradentate Phosphine and of a New Diphosphine Ether Mark R
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1991 Transition metal complexes of a novel tetradentate phosphine and of a new diphosphine ether Mark R. Mason Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Inorganic Chemistry Commons Recommended Citation Mason, Mark R., "Transition metal complexes of a novel tetradentate phosphine and of a new diphosphine ether " (1991). Retrospective Theses and Dissertations. 9554. https://lib.dr.iastate.edu/rtd/9554 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfîlm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from ary type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely afreet reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps.